


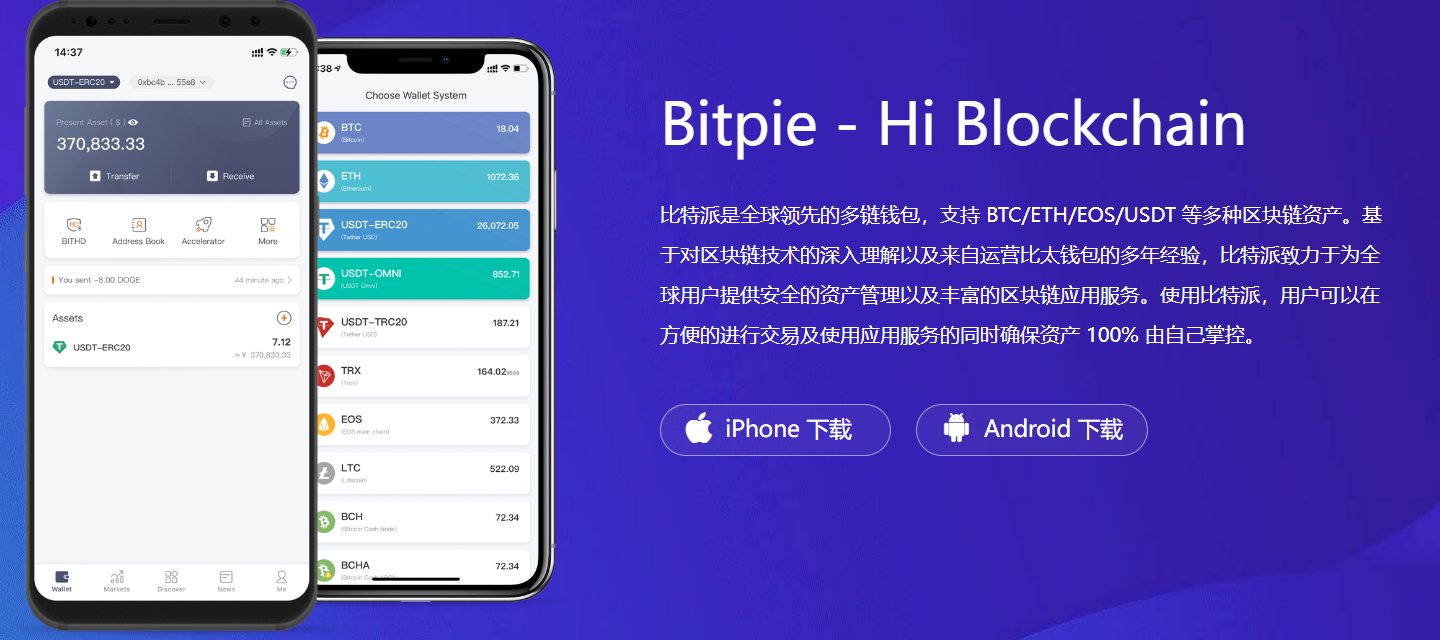
tokenpocket钱包官方苹果app下载|smr
- 作者: tokenpocket钱包官方苹果app下载
- 2024-03-07 21:09:30
SMR | Yang Lab
SMR | Yang Lab
SMR
Summary-data-based Mendelian Randomization
GCTA
SMR
GSMR
OSCA
CTG forum
Yang Lab
Loading...
If the website dose not work properly, you may need to update your Internet browser and enable javascript
or downlad offline PDF document, executable Files
Overview
About
The SMR software tool was originally developed to implement the SMR & HEIDI methods to test for pleiotropic association between the expression level of a gene and a complex trait of interest using summary-level data from GWAS and expression quantitative trait loci (eQTL) studies (Zhu et al. 2016 Nature Genetics). The SMR & HEIDI methodology can be interpreted as an analysis to test if the effect size of a SNP on the phenotype is mediated by gene expression. This tool can therefore be used to prioritize genes underlying GWAS hits for follow-up functional studies. The methods are applicable to all kinds of molecular QTL (xQTL) data, including DNA methylation QTL (mQTL) and protein abundance QTL (pQTL).
The SMR tool has subsequently been extended to include more analytical methods including SMR-multi (an extension of the SMR test to use multiple cis-xQTL SNPs; Wu et al. 2018 Nature Communications) and MeCS (a method for meta-analysis of xQTL data sets accounting for correlations among data sets; Qi et al. 2018 Nature Communications).
Credits
Futao Zhang developed original version of the
the software tool and webpages with supports from Zhili Zheng,
Zhihong Zhu,
Ting Qi,
Yang Wu,
and Jian Yang.
Zhihong Zhu
and Jian Yang
developed the SMR and HEIDI methods.
Ting Qi
and Jian Yang
developed the MeCS method.
Hailing Fang is currently maintaining the software.
Questions and Help Requests
Bug reports or questions to Jian Yang (jian.yang@westlake.edu.cn) at School of Life Sciences, Westlake University.
Citations
SMR & HEIDI methods and software tool
Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR,
Powell JE, Montgomery GW, Goddard ME, Wray NR, Visscher PM & Yang J (2016) Integration of summary data
from GWAS and eQTL studies predicts complex trait gene targets.
Nature Genetics, 48:481-487.
Multi-SNP-based SMR method and omic-data-based SMR analysis
Wu Y, Zeng J, Zhang F, Zhu Z, Qi T, Zheng Z, Lloyd-Jones LR, Marioni RE, Martin NG, Montgomery GW, Deary IJ, Wray NR, Visscher PM, McRae AF & Yang J (2018) Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nature Communications, 9: 918.
MeCS method
Qi T, Wu Y, Zeng J, Zhang F, Xue A, Jiang L, Zhu Z, Kemper K, Yengo L, Zheng Z, eQTLGen Consortium, Marioni RE, Montgomery GW, Deary IJ, Wray NR, Visscher PM, McRae AF & Yang J (2018) Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nature Communications, 9: 2282.
SMR & HEIDI analysis
SMR
# run SMR and HEIDI test
smr --bfile mydata --gwas-summary mygwas.ma --beqtl-summary myeqtl --out mysmr --thread-num 10
--bfile reads individual-level SNP genotype data (in PLINK
binary format) from a reference sample for LD estimation, i.e. .bed,
.bim, and .fam files.
--gwas-summary reads summary-level data from GWAS. The input
format follows that for GCTA-COJO analysis (
https://yanglab.westlake.edu.cn/software/gcta/#COJO).
smr --bld mybld --gwas-summary mygwas.ma --beqtl-summary myeqtl --out mysmr --thread-num 10
--bld reads LD information from a binary file in BLD format
mygwas.ma
SNP A1 A2 freq b se p n
rs1001 A G 0.8493 0.0024 0.0055 0.6653 129850
rs1002 C G 0.03606 0.0034 0.0115 0.7659 129799
rs1003 A C 0.5128 0.045 0.038 0.2319 129830
......
Columns are SNP, the coded allele (also called the effect allele or the reference allele), the other allele (also called the alternative allele), frequency of the effect allele, effect size, standard error, p-value and sample size. The headers are not keywords and will be omitted by the program. Important: “A1” needs to be the effect allele with “A2” being the other allele and “freq” needs to be the frequency of “A1”. NOTE:1) For a case-control study, the effect size should be log(odds ratio) with its corresponding standard error. 2) We use the GCTA-COJO format here to be compatible with the GCTA software. Note that the column "n" will not be used in either SMR or HEIDI analysis and thus can be replaced by "NA" if not available. 3) The allele frequency information in column "freq" will be used in a QC step to remove SNPs with discrepant allele frequencies between data sets.
--beqtl-summary reads summary-level data from a eQTL study in
binary format. We store eQTL summary data in three separate files
.esi (SNP information, in the same format as the PLINK .bim file),
.epi (probe information) and .besd (eQTL summary statistics in
binary format). See Data Management for more
information. We have prepared the data from the Westra study (Westra
et al. 2013 Nat Genet) in this format, which is available for
download at Download.
--out saves the results from the SMR analysis in .smr file
(text format).
mysmr.smr
ProbeID Probe_Chr Gene Probe_bp SNP SNP_Chr SNP_bp A1 A2 Freq b_GWAS se_GWAS p_GWAS b_eQTL se_eQTL p_eQTL b_SMR se_SMR p_SMR p_HEIDI nsnp_HEIDI
prb01 1 Gene1 1001 rs01 1 1011 C T 0.95 -0.024 0.0063 1.4e-04 0.36 0.048 6.4e-14 -0.0668 0.0197 6.8e-04 NA NA
prb02 1 Gene2 2001 rs02 1 2011 G C 0.0747 0.0034 0.0062 5.8e-01 0.62 0.0396 2e-55 0.0055 0.01 5.8e-01 4.17e-01 28
......
Columns are probe ID, probe chromosome, gene name, probe position, SNP name,SNP chromosome, SNP position, the effect (coded) allele, the other allele, frequency of the effect allele (estimated from the reference samples), effect size from GWAS, SE from GWAS, p-value from GWAS, effect size from eQTL study, SE from eQTL study, p-value from eQTL study, effect size from SMR, SE from SMR, p-value from SMR, p-value from HEIDI (HEterogeneity In Depedent Instruments) test, and number of SNPs used in the HEIDI test.
Missing Value is represented by "NA".
--thread-num specifies the number of OpenMP threads for
parallel computing. The default value is 1.
# Specify a method for HEIDI test
smr --bfile mydata --gwas-summary mygwas.ma --beqtl-summary myeqtl --heidi-mtd 0 --out mysmr
--heidi-mtd specifies a method for HEIDI test. 0 for the
original HEIDI test approach as in Zhu et al. (2016 Nature
Genetics),
and 1 for a new HEIDI test. The default
value is 1. The new approach uses up to the top 20 SNPs in the
cis-eQTL region (including the top cis-eQTL) for heterogeneity test
because our latest simulation shows that the power of HEIDI test
increases initially but then decreases with increasing number of
SNPs (m) with a peak at m = ~20.
# Filter SNPs by MAF (in the reference sample)
smr --bfile mydata --gwas-summary mygwas.ma --beqtl-summary myeqtl --maf 0.01 --out mysmr
--maf removes SNPs based on a minor allele frequency (MAF)
threshold in the reference sample.
# Specify a threshold to remove SNPs with discrepant allele frequencies between data sets
smr --bfile mydata --gwas-summary mygwas.ma --beqtl-summary myeqtl --diff-freq 0.1 --diff-freq-prop 0.05 --out mysmr
--diff-freq reads a threshold for allele frequency QC. That is, the SNPs with allele frequency differences between any pairwise data sets (including the LD reference sample, the eQTL summary data and the GWAS summary data) large than the specified threshold will be excluded. The default value is 0.2.
--diff-freq-prop reads a value as the maximum proportion of SNPs that are allowed to have allele frequency differences. The analysis will stop (with an error massage) if the proportion of SNPs being excluded by --diff-freq is larger than the specified value here. The default value is 0.05.
# Include or exclude a subset of individuals
smr --bfile mydata --gwas-summary mygwas.ma --beqtl-summary myeqtl --keep myindi.list --out mysmr
--keep includes a subset of individuals in the reference sample
for analysis.
--remove excludes a subset of individuals in the reference
sample from the analysis.
myindi.list
F001 S001
F002 S002
F003 S001
...
# Include or exclude a subset of eQTL summary data
smr --bfile mydata --gwas-summary mygwas.ma --beqtl-summary myeqtl --extract-snp mysnp.list --extract-probe myprobe.list --out mysmr
--extract-snp extracts a subset of SNPs for analysis.
--exclude-snp excludes a subset of SNPs from analysis.
mysnp.list
rs1001
rs1002
rs1003
...
--extract-probe extracts a subset of probes for analysis.
--exclude-probe excludes a subset of probes from analysis.
myprobe.list
probe1001
probe1002
probe1003
...
# Other parameters
smr --bfile mydata --gwas-summary mygwas.ma --beqtl-summary myeqtl --peqtl-smr 5e-8 --ld-upper-limit 0.9 --ld-lower-limit 0.05 --peqtl-heidi 1.57e-3 --heidi-min-m 3 --heidi-max-m 20 --cis-wind 2000 --thread-num 5 --out mysmr
--peqtl-smr p-value threshold to select the top associated eQTL
for the SMR test. The default value is 5.0e-8. By default, we only
run the SMR analysis in the cis regions. Please see below for the
SMR analysis in trans regions.
--peqtl-heidi threshold of eQTL p-value to select eQTLs for the
HEIDI test. The default value is 1.57e-3, which is equivalent to a
chi-squared value (df=1) of 10.
--ld-upper-limit LD r-squared threshold used to prune SNPs (eQTLs) in the
HEIDI test, i.e. removing SNPs in very strong LD with the top associated eQTL.
The default value is 0.9.
--ld-lower-limit LD r-squared threshold used to prune SNPs (eQTLs) in the
HEIDI test, i.e. removing SNPs in low LD or not in LD with the top associated eQTL.
The default value is 0.05.
--heidi-min-m minimum requirement of the number of cis-SNPs used in
the HEIDI test. We will skip the HEIDI test if the number of SNPs is
smaller than the threshold. This is because if the number of SNPs is
too small, HEIDI test has little power to detect heterogeneity and
possibly generates misleading result. The default value is 3.
--heidi-max-m maximum number of eQTLs used in
the HEIDI test. If the number of cis-SNPs included in the HEIDI test after LD pruning is larger than m, then only the top m SNPs (ranked by eQTL p-value) will be used in the test. The default value is 20.
--cis-wind defines a window centred around the probe to select
cis-eQTLs (passing a p-value threshold) for the SMR analysis. The
default value is 2000Kb.
# Specify a target SNP for the SMR and HEIDI tests
By default, we use the top cis-eQTL as a target in the SMR analysis,
i.e. using the top cis-eQTL in the SMR test and then using the top
cis-eQTL to test against the other cis-eQTLs in the region for
heterogeneity in the HEIDI test. You can also specific the target by the
following option. Note that this option will ignore p-value specified by
the --peqtl-smr option (--peqtl-heidi still applies).
smr --bfile mydata --gwas-summary mygwas.ma --beqtl-summary myeqtl --target-snp rs12345 --out mysmr
--target-snp specifies a SNP as the target for the SMR and
HEIDI tests as described above.
# Specify a list of SNP-probe pairs for the SMR and HEIDI tests
Here we provide two flags to run a batch of SMR analyses based on a user-specified list of SNP-probe pairs. For each probe, the program will only read the data of SNPs within +/- 2000Kb (can be changed by the --smr-wind flag) of the specified SNP. Note that the SNP can be distant from the corresponding probe.
smr --bfile mydata --gwas-summary mygwas.ma --beqtl-summary myeqtl --extract-snp-probe snp_probe.list --out mysmr
--extract-snp-probe specifies a SNP-probe list (see the format below).
smr --bfile mydata --gwas-summary mygwas.ma --beqtl-summary myeqtl --extract-target-snp-probe snp_probe.list --out mysmr
--extract-target-snp-probe specifies a SNP-probe list and forces the specified SNP (not necessarily being the top associated SNP) as the target SNP for the SMR and HEIDI tests.
snp_probe.list
rs1001 probe1001
rs1002 probe1002
rs1002 probe1003
...
# Turn off the HEIDI test
smr --bfile mydata --gwas-summary mygwas.ma --beqtl-summary myeqtl --heidi-off --out mysmr
--heidi-off turns off the HEIDI test.
SMR and HEIDI tests in trans regions
The trans-eQTLs are defined as the eQTLs that are more than 5Mb away
from the probe.
smr --bfile mydata --gwas-summary mygwas.ma --beqtl-summary myeqtl --out mytrans --trans --trans-wind 1000
--trans turns on SMR and HEIDI tests in trans regions.
--trans-wind defines the size of a window on either side of the top associated
trans-eQTL to select SNPs (passing a p-value threshold) for the SMR
and HEIDI test. The default value is 1000 Kb (i.e. a whole region of 2000 Kb).
mytrans.smr
ProbeID Probe_Chr Gene Probe_bp trans_chr trans_leftBound trans_rightBound SNP SNP_Chr SNP_bp A1 A2 Freq b_GWAS se_GWAS p_GWAS b_eQTL se_eQTL p_eQTL b_SMR se_SMR p_SMR p_HEIDI nsnp_HEIDI
prb01 1 Gene1 1001 16 5349752 7350902 rs01 16 6349942 C T 0.131 0.0021 0.0152 8.8e-01 -0.214 0.038 3.26e-08 -0.0098 0.071 8.9e-01 1.73e-1 19
prb01 1 Gene1 1001 21 6443018 8459725 rs02 21 7460164 G C 0.0747 0.0034 0.0062 5.8e-01 0.62 0.0396 2e-55 0.0055 0.01 5.8e-01 4.17e-01 8
......
Columns are probe ID, probe chromosome, gene name, probe position,
tans-eQTL chromosome, left boundary of the trans-region, right boundary
of the trans-region, SNP name, SNP chromosome, SNP position, the effect
(coded) allele, the other allele, frequency of the effect allele
(estimated from the reference samples), effect size from GWAS, SE from
GWAS, p-value from GWAS, effect size from eQTL study, SE from eQTL
study, p-value from eQTL study, effect size from SMR, SE from SMR,
p-value from SMR, p-value from HEIDI test, and number of SNPs used in
the HEIDI test.
Multi-SNP-based SMR test
Below shows an option to combine the information from all the SNPs in a
region that pass a p-value threshold (the default value is 5.0e-8 which
can be modified by the flag --peqtl-smr) to conduct a multi-SNP-based SMR
analysis (Wu et al. 2018 Nature Communications).
The SNPs are pruned for LD using a weighted vertex coverage algorithm
with a LD r2 threshold (the default value is 0.9 which can be modified
by the flag --ld-pruning) and eQTL p-value as the weight.
smr --bfile mydata --gwas-summary mygwas.ma --beqtl-summary myeqtl --out mymulti --smr-multi
--smr-multi turns on set-based SMR test in the cis-region.
smr --bfile mydata --gwas-summary mygwas.ma --beqtl-summary myeqtl --out mymulti --smr-multi --set-wind 500
--set-wind defines a window width (Kb) centred around the top
associated cis-eQTL to select SNPs in the cis-region. The default
value is -9 resulting in selecting SNPs in the whole cis-region if
this option is not specified.
smr --bfile mydata --gwas-summary mygwas.ma --beqtl-summary myeqtl --out mymulti --smr-multi --ld-multi-snp 0.1
--ld-multi-snp LD r-squared threshold used to prune SNPs (eQTLs) in the Multi-SNP based SMR test. The default value is 0.1.
SMR analysis of two molecular traits
Here we provide an option to test the pleotropic association between two
molecular traits using summary data. Take the analysis of DNA
methylation and gene expression data as an example. In this case, we
will need mQTL and eQTL summary data in BESD format. The current version of the program focuses only on the analysis
in the cis-region, i.e. only testing for associations between genes and DNA methylation sites that are in less than 2 Mb distance.
smr --bfile mydata --beqtl-summary myexposure --beqtl-summary myoutcome --out myomics
--beqtl-summary the first one reads mQTL summary data as the
exposure. The second one reads eQTL summary data from as the
outcome.
smr --bfile mydata --beqtl-summary myexposure --beqtl-summary myoutcome --extract-exposure-probe myeprobein.list --out myomics
--extract-exposure-probe extracts a subset of exposure probes
for analysis.
--extract-outcome-probe extracts a subset of outcome probes for
analysis.
--exclude-exposure-probe excludes a subset of exposure probes
from analysis.
--exclude-outcome-probe excludes a subset of outcome probes
from analysis.
smr --bfile mydata --beqtl-summary myexposure --beqtl-summary myoutcome --extract-single-exposure-probe eprobe1 --extract-single-outcome-probe oprobe1 --out myomics
--extract-single-exposure-probe extracts a single exposure
probe for analysis.
--extract-single-outcome-probe extracts a single outcome probe
for analysis.
smr --bfile mydata --beqtl-summary myexposure --beqtl-summary myoutcome --exclude-single-exposure-probe eprobe1 --exclude-single-outcome-probe oprobe1 --out myomics
--exclude-single-outcome-probe excludes a single outcome probe
from analysis.
--exclude-single-exposure-probe excludes a single exposure
probe from analysis.
Data Management
eQTL summary data are usually generated from association tools such as
PLINK and stored in separate files in text format (usually one file for
each probe) with a very large file size in total. Here we provide an
efficient way to store the eQTL summary data in binary format (BESD),
with flexible options to query the data for any subset of SNPs and/or
probes (see Query eQTL Results). We further provide
a sparse version of the BESD format (used as default), which is
extremely storage-efficient without losing too much information. The
basic idea is that we store summary data of all SNPs within 2Mb of a
probe in either direction, all SNPs within 1Mb of any trans-eQTL in
either direction, and all SNPs with p < 1e-5 in the rest of the genome
(note that all these parameters can be specified by users). We also
provide several options to import data in various of formats (e.g.
PLINK, GEMMA, BOLT-LMM and other text formats).
BESD format
# BESD format: an efficient format to store eQTL summary data
We store eQTL summary data in three separate files .esi (SNP
information, similar as a PLINK .bim file), .epi (probe information) and
.besd (a binary file to store the summary statistics).
myeqtl.esi
1 rs1001 0 744055 A G 0.23
1 rs1002 0 765522 C G 0.06
1 rs1003 0 995669 T C 0.11
......
Columns are chromosome, SNP, genetic distance (can be any arbitary value
since it will not be used in the SMR analysis), basepair position, the
effect (coded) allele, the other allele and frequency of the effect
allele.
myeqtl.epi
1 probe1001 0 924243 Gene01 +
1 probe1002 0 939564 Gene02 -
1 probe1003 0 1130681 Gene03 -
......
Columns are chromosome, probe ID(can be the ID of an exon or a
transcript for RNA-seq data), genetic distance (can be any arbitary
value), physical position, gene ID and gene orientation (this
information will only be used for graphic presentation, please see [SMR plot]).
myeqtl.besd
eQTL summary-level statistics (effect size and SE) in binary format.
Please do not try to open this file with a text editor.
Given the large numbers of SNPs and probes, the size of a .besd file
will still be very large. Since the eQTL association signals are highly
enriched in the cis-region and often there are not many trans-eQTLs, we
could reduce the size of the .besd file by orders of magnitude if we
only store the data for SNPs within 2Mb of a probe in either direction,
SNPs within 1Mb of any trans-eQTL in either direction, and SNPs with p < 1e-5
in the rest of the genome (see below for options to change these
paramters). We call this the sparse BESD format.
We only store effect size (b) and SE in the BESD file, and re-calculate
p-value for analysis when necessary, assuming b / SE follows a standard
normal distribution, i.e. N(0, 1). Strictly speaking, b / SE follows a
t-distribution which is approximately N(0, 1) if sample size is large.
For data sets with small sample sizes (e.g. GTEx), this might lead to a bias
in p-value. In this scenario, we recommend users to compute z* based on the original p-value from
a standard normal distribution, and adjust the standard error as SE = b
/ z*. This adjustment guarantees that the re-computed p-value from b
and SE being exact the same as the original p-value.
See below for options to make a BESD file from data in several different
formats.
Make a BESD file
We provide eight different ways of converting cis-eQTL data in other formats to BESD format.
1. Make a BESD file from eQTL summary data in ESD format
To compile data in sparse BESD format
smr --eqtl-flist my.flist --make-besd --out mybesd
--eqtl-flist reads a file to get probe information and file
paths of the eQTL summary data.
--make-besd saves summary data in BESD format. By default, the
data will be stored in sparse BESD format (See below for the option
--make-besd-dense to store the data in dense BESD format). By
default, the data will be stored in sparse BESD format if the
sparsity given the parameters (by default, ±2Mb of the cis-region,
±1Mb of any trans-eQTL and all SNP at p < 1e-5) is lower than 0.4. It
will also output a text file (.summary) to summarise the genomic
regions stored in the .besd file (sparse format) for each probe.
my.flist
Chr ProbeID GeneticDistance ProbeBp Gene Orientation PathOfEsd
9 cg00000658 0 139997924 MAN1B1 - path/my01.esd
20 cg26036652 0 33735834 NA NA path/my02.esd
1 cg00489772 0 3775078 NA NA path/my03.esd
......
This is a text file with headers. The first 6 columns are the same as in .epi. The last column is the full path of an eQTL summary data file (.esd file, see below for the format of a .esd file).
my01.esd
Chr SNP Bp A1 A2 Freq Beta se p
9 rs12349815 150048 T A 0.968 0.019 0.016 0.2434
20 rs141129176 62955484 G A 0.89 0.012 0.009 0.2156
......
This is a text file with headers. Columns are chromosome, SNP, the
effect (coded) allele, the other allele, basepair position, frequency of
the effect allele, effect size, standard error and p-value.
HINT : if the SNPs in all of the .esd files are identical, the
efficiency of the analysis can be largely improved by adding the
--geno-uni option. This option call be used in all the commands of this
section.
smr --eqtl-flist my.flist --make-besd --geno-uni --out mybesd
--geno-uni indicates all the input .esd files are identical.
To compile eQTL summary data in sparse BESD format with user-specified
parameters
smr --eqtl-flist my.flist --make-besd --cis-wind 2000 --trans-wind 1000 --peqtl-trans 5.0e-8 --peqtl-other 1.0e-5 --out mybesd
--cis-wind specifies a window (in Kb unit) to store all the
SNPs within the window of the probe in either direction. The default
value is 2000Kb.
--trans-wind specifies a window (in Kb unit) to store all the
SNPs in a trans-region. If there is a trans-eQTL with p-value < the
specified threshold (--peqtl-trans), it will store all the SNPs
within the window of the top associated trans-eQTL in either
direction. The default value is 1000Kb.
--peqtl-trans p-value threshold for trans-eQTLs. The default
value is 5.0e-8.
--peqtl-other Apart from the cis and trans regions, it will
also store all SNPs with eQTL p-values < this threshold. The
default value is 1.0e-5 .
To compile the eQTL summary data in dense BESD format
smr --eqtl-flist my.flist --make-besd-dense --out mybesd
--make-besd-dense saves summary data of all SNPs for all probes.
WARNING : This will generate a huge file.
NOTE : the --make-besd-dense option can be used in all the commands above and below.
2. Make a BESD file from Matrix eQTL output
smr --eqtl-summary mateQTL.txt --matrix-eqtl-format --make-besd --out mybesd
--eqtl-summary reads eQTL summary statistics in text format or compressed text format (e.g. *.tar.gz file).
--matrix-eqtl-format indicates eQTL summary data in Matrix eQTL output format.
mateQTL.txt
SNP gene beta t-stat p-value FDR
rs13258200 ENSG00000071894.10 -1.00783189089702 -16.641554315712 2.3556801409867e-24 1.12905157909337e-18
rs6599528 ENSG00000071894.10 -1.06253739134798 -15.8412867110849 2.73027622294589e-23 5.51886367106636e-18
rs2272666 ENSG00000071894.10 -1.04810713295621 -15.6736668186937 4.6058755123246e-23 5.51886367106636e-18
rs4313195 ENSG00000071894.10 -1.04810713295621 -15.6736668186937 4.6058755123246e-23 5.51886367106636e-18
rs2280836 ENSG00000071894.10 -1.00773805984667 -15.2332537951202 1.84980830591084e-22 1.77318554626341e-17
...
This file has headers. The six columns are SNP, gene, beta (i.e. SNP effect on gene expression), t-statistic, p-value and q-value
([http://www.bios.unc.edu/research/genomic_software/Matrix_eQTL/]).
NOTE : 1) The program is able to read *.tar.gz file. 2) The SNP and probe information in the SMR eQTL output files (.esi and .epi) converted from Matrix eQTL output are not complete and need to be updated using the options in Update a BESD file.
3. Make a BESD file from FastQTL output
smr --eqtl-summary fastqtlnomi.txt --fastqtl-nominal-format --make-besd --out mybesd
--fastqtl-nominal-format indicates eQTL summary data in FastQTL "nominal pass" output format.
fastqtlnomi.txt
ENSG00000237438.1 indel:4D_22_16518157 -999303 0.542909 -0.0510761
ENSG00000237438.1 snp_22_16519289 -998171 0.432764 0.124424
ENSG00000237438.1 snp_22_16520141 -997319 0.0945196 -0.251906
ENSG00000237438.1 snp_22_16520948 -996512 0.102846 -0.274157
ENSG00000237438.1 snp_22_16523696 -993764 0.0676318 -0.324492
ENSG00000237438.1 snp_22_16523730 -993730 0.0674215 -0.206578
...
This file has no header. the five columns are gene, SNP, distance in bp between the SNP and the gene, p-value and beta (i.e. SNP effect on gene expression)
([http://fastqtl.sourceforge.net/pages/cis_nominal.html]).
NOTE : 1) The FastQTL output file should be generated by a FastQTL version higher than v2.184. 2) The program is able to read *.tar.gz file. 3) The SNP and probe information in the SMR output files (.esi and .epi) converted from FastQTL output are not complete and need to be updated using the options in Update a BESD file.
4. Make a BESD file from eQTL summary data in PLINK-qassoc output format
The output file from a PLINK --assoc analysis does not contain allele
information. We therefore need to read the alleles from a PLINK .bim
file. The file path of the PLINK .bim file needs to be added as the last
column of the .flist file (see the example below).
smr --eqtl-flist my.flist --plink-qassoc-format --make-besd --out mybesd
--plink-qassoc-format reads eQTL summary data in PLINK-qassoc output format (output file from a PLINK --assoc analysis for a quantitative trait).
my.flist
Chr ProbeID GeneticDistance ProbeBp Gene Orientation PathOfEsd PathOfBim
9 cg00000658 0 139997924 MAN1B1 - path_assoc/my01.qassoc path_genotype/chr9
20 cg26036652 0 33735834 NA NA path_assoc/my02.qassoc path_genotype/chr20
1 cg00489772 0 3775078 NA NA path_assoc/my03.qassoc path_genotype/chr19
......
NOTE : The program is able to read *.tar.gz file, e.g.
path_assoc/my03.qassoc.tar.gz
5. Make a BESD file from eQTL summary data in GEMMA output format
smr --eqtl-flist my.flist --gemma-format --make-besd --out mybesd
--gemma-format reads eQTL summary data in GEMMA association output format
chr rs ps n_miss allel1 allel0 af beta se l_remle p_wald
1 rs3683945 3197400 0 A G 0.443 -7.788665e-02 6.193502e-02 4.317993e+00 2.087616e-01
1 rs3707673 3407393 0 G A 0.443 -6.654282e-02 6.210234e-02 4.316144e+00 2.841271e-01
1 rs6269442 3492195 0 A G 0.365 -5.344241e-02 5.377464e-02 4.323611e+00 3.204804e-01
......
The 11 columns are: chromosome, SNP ID, basepair position, number of
missing values for a given SNP, the effect (coded) allele, the other
allele, frequency of the effect allele, effect size, standard error,
lambda and p-value
([http://www.xzlab.org/software.html]).
6. Make a BESD file from eQTL summary data in BOLT-LMM output format
smr --eqtl-flist my.flist --bolt-assoc-format --make-besd --out mybesd
--bolt-assoc-format reads eQTL summary data in BOLT_LMM output
format
SNP CHR BP GENPOS ALLELE1 ALLELE0 A1FREQ F_MISS BETA SE P_BOLT_LMM_INF P_BOLT_LMM
rs58108140 1 10583 0.000000 A G 0.109810 0.011935 0.074942 0.045043 9.6E-02 9.7E-02
rs180734498 1 13302 0.000000 T C 0.061042 0.007595 0.084552 0.058078 1.5E-01 1.4E-01
rs151118460 1 91581 0.000000 A G 0.399377 0.013382 0.024344 0.034394 4.8E-01 4.8E-01
......
The 12 columns are: SNP ID, chromosome, basepair position, genetic
position, the effect (coded) allele, the other allele, frequency of the
effect allele, fraction of individuals with missing genotype at the SNP,
effect size, standard error, infinitesimal model (mixture model)
association test p-value, and non-infinitesimal model association test
p-value
([https://data.broadinstitute.org/alkesgroup/BOLT-LMM/#x1-440008.1]).
7. Make a BESD file from SMR query output
smr --qfile myquery.txt --make-besd --out mybesd
--qfile reads eQTL summary data in SMR query output format (see
Query eQTL Results for the format of a query output
file).
8. Make a BESD file from BESD file(s)
To make a sparse BESD file from a single dense BESD file
smr --beqtl-summary my_beqtl --make-besd --out my_sparse
smr --beqtl-summary my_beqtl --cis-wind 2000 --trans-wind 1000 --peqtl-trans 5.0e-8 --peqtl-other 1.0e-5 --make-besd --out my_sparse
To make a sparse BESD file from multiple sparse or dense BESD files (can
be a mixture of both types)
smr --besd-flist my_file.list --make-besd --out my_sparse
--besd-flist reads a file to get the full paths of the BESD
files.
my_file.list
path1/my_besd1
path2/my_besd2
path3/my_besd3
...
NOTE : this command can be used to merge multiple BESD files.
HINT : if the SNPs in all the .esi files are identical, you can speed up
the analysis using the --geno-uni option.
9. Make a BESD file from QTLtools output
smr --eqtl-summary qtltoolsnomi.txt --qtltools-nominal-format --make-besd --out mybesd
--qtltools-nominal-format indicates eQTL summary data in QTLtools "nominal pass" output format.
qtltoolsnomi.txt
ENSG00000237438.2 chr22 17517460 17517460 + 5803 -165953 rs5748687 chr22 17351507 17351507 0.00227542 -0.225077 0
ENSG00000237438.2 chr22 17517460 17517460 + 5803 -165363 rs4006343 chr22 17352097 17352097 0.0022876 -0.226253 0
ENSG00000237438.2 chr22 17517460 17517460 + 5803 -164231 rs3875996 chr22 17353229 17353229 0.00181073 -0.233649 0
ENSG00000099954.14 chr22 17840837 17840837 + 6200 469530 rs5992121 chr22 18310367 18310367 0.00653855 -0.00519037 0
ENSG00000099954.14 chr22 17840837 17840837 + 6200 473554 rs367922 chr22 18314391 18314391 0.00125305 -0.00612444 0
ENSG00000099954.14 chr22 17840837 17840837 + 6200 476586 rs11705197 chr22 18317423 18317423 0.00961104 -0.00498535 0
...
This file has no header. the 14 columns are The columns are: 1. The phenotype ID 2. The chromosome ID of the phenotype 3. The start position of the phenotype 4. The end position of the phenotype 5. The strand orientation of the phenotype 6. The total number of variants tested in cis 7. The distance between the phenotype and the tested variant (accounting for strand orientation) 8. The ID of the tested variant 9. The chromosome ID of the variant 10. The start position of the variant 11. The end position of the variant 12. The nominal P-value of association between the variant and the phenotype 13. The corresponding regression slope 14. A binary flag equal to 1 is the variant is the top variant in cis.
([https://qtltools.github.io/qtltools/pages/modecisnominal.html]).
smr --eqtl-summary qtltoolspermu.txt --qtltools-permu-format --make-besd --out mybesd
--qtltools-permu-format indicates eQTL summary data in QTLtools "permutaion pass" output format.
qtltoolspermu.txt
ENSG00000273442.1 chr22 17561591 17561591 + 5911 -8065 rs73149812 chr22 17553526 17553526 356 308.754 1.08518 661.618 0.000244522 0.134045 0.318681 0.304744
ENSG00000177663.9 chr22 17565844 17565844 + 5919 -52141 rs2908526 chr22 17513703 17513703 356 333.628 0.998034 982.885 8.03358e-14 -0.209617 0.000999001 4.92276e-10
ENSG00000183307.3 chr22 17602257 17602257 - 5967 14282 rs6518661 chr22 17587975 17587975 356 335.297 0.983505 921.283 1.56789e-07 -0.00776053 0.000999001 0.00038022
ENSG00000069998.8 chr22 17646177 17646177 - 6044 3400 rs71200232 chr22 17642776 17642777 356 332.081 1.03052 958.093 8.87771e-17 -1.49798 0.000999001 3.8205e-13
ENSG00000093072.11 chr22 17702879 17702879 - 6048 3249 rs5747027 chr22 17699630 17699630 356 287.216 1.02518 511.292 0.000578649 0.640374 0.628372 0.629492
...
This file has no header. the 19 columns are The columns are: 1. The phenotype ID 2. The chromosome ID of the phenotype 3. The start position of the phenotype 4. The end position of the phenotype 5. The strand orientation of the phenotype 6. The total number of variants tested in cis 7. The distance between the phenotype and the tested variant (accounting for strand orientation) 8. The ID of the top variant 9. The chromosome ID of the top variant 10. The start position of the top variant 11. The end position of the top variant 12. The number of degrees of freedom used to compute the P-values 13. Dummy 14. The first parameter value of the fitted beta distribution 15. The second parameter value of the fitted beta distribution (it also gives the effective number of independent tests in the region) 16. The nominal P-value of association between the phenotype and the top variant in cis 17. The corresponding regression slope 18. The P-value of association adjusted for the number of variants tested in cis given by the direct method (i.e. empirircal P-value) 19. The P-value of association adjusted for the number of variants tested in cis given by the fitted beta distribution. We strongly recommend to use this adjusted P-value in any downstream analysis
([https://qtltools.github.io/qtltools/pages/modecispermutation.html]).
NOTE : 1) The program is able to read *.tar.gz file. 2) The SNP and probe information in the SMR eQTL output files (.esi and .epi) converted from QTLtools output are not complete and need to be updated using the options in Update a BESD file.
Update a BESD file
Some of the information such as SNP chromosome, SNP position, the effect allele, the other allele, probe chromosome and probe position might be missing in the output files generated by some of the eQTL analysis tools (e.g. Matrix eQTL and FastQTL). These information are necessary for the SMR analysis and some of the data management operations in the SMR tool, and thus need to be updated in the .esi or .epi file.
Users can update the .esi file and .epi file manually, but please be aware of not changing the order of SNPs in the .esi file or probes in the .epi file because they are associated with the information in the .besd file. We also provide the options below to update the information in the .esi and .epi files.
# Update the .esi or .epi files
smr --beqtl-summary my_beqtl --update-esi mybigpool.esi
--update-esi reads a .esi file for updating and backup the original .esi file.
smr --beqtl-summary my_beqtl --update-epi mybigpool.epi
--update-epi reads a .epi file for updating and backup the original .epi file.
# Add or update the frequencies of the effect alleles
smr --beqtl-summary myeqtl --update-freq mysnp.freq
--update-freq reads an input file with allele frequency
information and adds a new column (i.e. frequency the effect allele)
to the .esi file.
mysnp.freq
rs12349815 T A 0.968
rs141129176 G A 0.89
......
The input is a text file without headers. Columns are SNP, the
effect allele, the other allele and frequency of the effect allele.
NOTE : the SMR program is compatible with .esi files with or without
frequency information.
Mange the sample size
# Add or update the sample size in the BESD file
smr --beqtl-summary myeqtl --add-n 1000 --make-besd --out mybesd
--add-n reads the sample size.
NOTE : The flag is valid for all the options to make a BESD file. For example:
smr --qfile myquery.txt --add-n 100 --make-besd --out mybesd
# Show the sample size
smr --beqtl-summary myeqtl --show-n
--show-n shows the sample size on the screen or in the log output.
Extract/remove a subset of data
# Extract a subset of SNPs and/or probes
smr --beqtl-summary myeqtl --extract-snp mysnp.list --extract-probe myprobe.list --make-besd --out mybesd
# Remove a subset of SNPs and/or probes
smr --beqtl-summary myeqtl --exclude-snp mysnp.list --exclude-probe myprobe.list --make-besd --out mybesd
# Extract a subset of SNPs with p < a threshold
smr --beqtl-summary myeqtl --extract-snp-p 1e-5 --make-besd --out mybesd
--extract-snp-p reads a p-value threshold to extract SNPs
# Remove a subset of SNPs with p < a threshold
smr --beqtl-summary myeqtl --exclude-snp-p 1e-5 --make-besd --out mybesd
--exclude-snp-p reads a p-value threshold to exclude SNPs
# Extract cis-regions of eQTL summary data
smr --beqtl-summary myeqtl --extract-cis --make-besd --out mybesd
--extract-cis extracts the cis-eQTL summary data.
BLD format
We store LD information in two separate files .esi (SNP information, similar as a PLINK .bim file) and .bld (a binary file to store LD correlations).
# Make a BLD file from SNP genotype data.
smr --bfile mydata --make-bld --r --ld-wind 4000 --out mybld
--make-bld saves the LD correlations between SNPs in binary format.
--r calculates the LD correlations.
--ld-wind specifies a window in Kb unit to select SNP pairs for the LD computation. The default value is 4000 Kb.
To specify a chromosome for the LD computation
smr --bfile mydata --make-bld --r --ld-wind 4000 --chr 21 --out mybld
To specify a SNP as an anchor for the LD computation (computing the LD correlations between the anchor SNPs and other SNPs in a window)
smr --bfile mydata --make-bld --r --ld-wind 4000 --snp rs123 --out mybld
# Query a BLD file.
smr --query --bld mybld --out myld
--bld specifies a BLD file.
To specify a chromosome to query the LD information
smr --query --bld mybld --chr 21 --out myld
To query the LD between a specified SNP and other SNPs in a window
smr --query --bld mybld --snp rs123 --out myld.ld
smr --query --bld mybld --snp rs123 --ld-wind 2000 --out myld.ld
myld.ld
CHR_A BP_A SNP_A CHR_B BP_B SNP_B R
21 13446559 rs36061596 21 14571990 rs2822531 -0.000768446
21 13517135 rs2847443 21 14571990 rs2822531 0.00351777
21 13523286 rs2775537 21 14571990 rs2822531 -0.0312363
......
Remove technical eQTLs
# Remove technical eQTLs
Filtering out eQTLs for which there is a significant cis-eQTL in the
hybridization region of the probe. This option will remove all the SNPs
in the cis-region of the probe and save the removed data in a file in
SMR Query format (see Query eQTL Results for the format of a query
output file). The default p-value threshold is 5e-8,
which can be changed by the --p-technical (see below).
smr --beqtl-summary myeqtl --rm-technical probe_hybrid.txt --make-besd --out mybesd
--rm-technical specifies the probe hybridization region and
excludes the technical eQTLs.
probe_hybrid.txt
19 probe0 50310094 50310143
19 probe1 406496 406545
10 probe2 119293020 119293069
......
This is a text file without headers. Columns are chromosome, probe
ID, start of the hybridization region and end of the hybridization
region.
smr --beqtl-summary myeqtl --rm-technical probe_hybrid.txt --p-technical 5e-8 --make-besd --out mybesd
--p-technical reads a p-value threshold to select technical
eQTLs. The default value is 5e-8.
SMR locus plot
SMR locus plot
Here we provide an R script to plot SMR results as presented in Zhu et
al. (2016 Nature Genetics).
The data file for plot can be generated by the command below. The R
script is avaliable at Download.
# SMR command line to generate a data file for plot
smr --bfile mydata --gwas-summary mygwas.ma --beqtl-summary myeqtl --out myplot --plot --probe ILMN_123 --probe-wind 500 --gene-list glist-hg19
--plot saves the data file for plot in text format .
--gene-list specifies a gene range list.
glist-hg19 (without strand information)
19 58858171 58864865 A1BG
19 58863335 58866549 A1BG-AS1
10 52559168 52645435 A1CF
......
This is a text file without headers. The columns are chromosome code,
start of gene, end of gene and gene ID.
The gene range lists ( mirrored from
[PLINK2]
website) are
hg18:
glist-hg18
(older, ASCII-sorted PNGU
version)
hg19:
glist-hg19
hg38:
glist-hg38
Not all the genes in the region but only those in the .epi file will be
drawn in the locus plot since the strand information is absent from the
gene list provied in the PLINK2 website. To fix this problem, we updated
SMR to read a gene list with/without strand information.
glist-hg19 (with strand information, you need generate the list youself)
19 58858171 58864865 A1BG -
19 58863335 58866549 A1BG-AS1 +
10 52559168 52645435 A1CF -
......
This is a text file without headers. The columns are chromosome code,
start of gene, end of gene, gene ID and gene strand.
# R commands to draw the plots
source("plot_SMR.r")
# Read the data file in R:
SMRData = ReadSMRData("myplot.ILMN_123.txt")
# Plot the SMR results in a genomic region centred around a probe:
SMRLocusPlot(data=SMRData, smr_thresh=8.4e-6, heidi_thresh=0.05, plotWindow=1000, max_anno_probe=16)
# smr_thresh: genome-wide significance level for the SMR test.
# heidi_thresh: threshold for the HEIDI test. The default value is 0.05.
# cis_wind: size of a window centred around the probe to select cis-eQTLs for plot. The default value is 2000Kb.
# max_anno_probe: maximum number of probe names to be displayed on the figure. The default value is 16.
# Plot effect sizes from GWAS against those from eQTL study:
SMREffectPlot(data=SMRData, trait_name="BMI")
# trait_name: name of the trait or disease.
Omics SMR plot
A web-based tool to generate omics SMR plots (see the example below).
Query eQTL Results
Query eQTL Summary Results
Since the eQTL summary are stored in binary format for a large number of
probes and SNPs, we provide the options below to query the eQTL summary
results from command line options or from file-list options given a
specified eQTL p-value threshold.
# Command line options for SNPs
To query the eQTL resutls for a single SNP, we could use this command
smr --beqtl-summary myeqtl --query 5.0e-8 --snp rs123 --out myquery
--query saves in text format a subset of the eQTL summary
dataset based on the specified eQTL p-value threshold. The default
value is 5.0e-8.
--snp specifies a single SNP.
myquery.txt
SNP Chr BP A1 A2 Freq Probe Probe_Chr Probe_bp Gene Orientation b se p
rs01 1 1001 A G 0.23 cg01 1 1101 gene1 + -0.033 0.006 3.8e-08
rs01 1 1001 A G 0.06 cg02 1 1201 gene2 - 0.043 0.007 8.1e-10
......
To query eQTL resutls for a range of SNPs in a genomic region
smr --beqtl-summary myeqtl --query 5.0e-8 --from-snp rs123 --to-snp rs456 --out myquery
--from-snp specifies the start SNP.
--to-snp specifies the end SNP.
NOTE : All SNPs should be on the same chromosome.
To query eQTL results for all SNP on a chromosome
smr --beqtl-summary myeqtl --query 5.0e-8 --snp-chr 1
--snp-chr specifies a chromosome to select SNPs.
NOTE : The probes in the result could be on the other chromosomes if
there are trans-eQTLs.
To query SNPs based on physical positions
smr --beqtl-summary myeqtl --query 5.0e-8 --snp-chr 1 --from-snp-kb 100 --to-snp-kb 200 --out myquery
--from-snp-kb specifies the start physical position of the
region.
--to-snp-kb specifies the end physical position of the region.
NOTE : You will need to specify a chromosome (using the '--snp-chr'
option) when using this option.
To query based on a flanking region of a SNP
smr --beqtl-summary myeqtl --query 5.0e-8 --snp rs123 --snp-wind 50 --out myquery
--snp-wind defines a window (in Kb unit) centred on a specified SNP.
# Command line options for probes
To query based on a single probe
smr --beqtl-summary myeqtl --query 5.0e-8 --probe cg123 --out myquery
--probe specifies a single probe.
To query based on a range of probes
smr --beqtl-summary myeqtl --query 5.0e-8 --from-probe cg123 --to-probe cg456 --out myquery
--from-probe specifies the start probe.
--to-probe specifies the end probe.
NOTE : All probes should be on the same chromosome.
To query based on a chromosome
smr --beqtl-summary myeqtl --query 5.0e-8 --probe-chr 1
--probe-chr specifies a chromosome to select probes.
NOTE : The SNPs in the result could be on the other chromosomes if there
are trans-eQTLs.
To query based on physical positions of the probes
smr --beqtl-summary myeqtl --query 5.0e-8 --probe-chr 1 --from-probe-kb 1000 --to-probe-kb 2000 --out myquery
--from-probe-kb specifies the start physical position of the
probes.
--to-probe-kb specifies the end physical position of the
probes.
NOTE : You will need to specify a chromosome (using the '--probe-chr'
option) when using this option.
To query based on a flanking region of a probe
smr --beqtl-summary myeqtl --query 5.0e-8 --probe cg123 --probe-wind 1000 --out myquery
--probe-wind defines a window (in Kb unit) centred on a specified probe.
To query based on a gene
smr --beqtl-summary myeqtl --query 5.0e-8 --gene gene1 --out myquery
--gene specifies a single gene to select probes.
# Command line option for cis-region
smr --beqtl-summary myeqtl --query 5.0e-8 --probe cg123 --cis-wind 2000 --out myquery
# File-list options
To query based on a list of SNPs
smr --beqtl-summary myeqtl --extract-snp snp.list --query 5.0e-8 --out myquery
To query based on a list of probes
smr --beqtl-summary myeqtl --extract-probe probe.list --query 5.0e-8 --out myquery
To qurey based on a list of genes
smr --beqtl-summary myeqtl --genes gene.list --query 5.0e-8 --out myquery
--genes extracts a subset of probes which tag the genes in the
list.
gene.list
gene1
gene2
gene3
...
Descriptive summary of the cis-eQTL data
smr --descriptive-cis --beqtl-summary myeqtl --out mydescriptive
--descriptive-cis outputs the descriptive summary of the cis-eQTL data (including the number of probes, the boundaries of the each cis-region, the top cis-eQTL in each cis-eQTL region and the summary statistics of the top cis-eQTL) at p-value threshold (the default value is 5.0e-8; can be changed by --peqtl-cis). The size of the cis-region can be changed by --cis-wind (the default value is 2000Kb).
smr --descriptive-cis --beqtl-summary myeqtl --cis-wind 2000 --out mydescriptive
Descriptive summary of the trans-eQTL data
smr --descriptive-trans --beqtl-summary myeqtl --out mydescriptive
--descriptive-trans outputs the descriptive summary of the trans-eQTL data at p-value threshold (the default value is 5.0e-8; can be changed by --peqtl-trans). The size of a trans-region can be changed by --trans-wind (the default value is 1000Kb). Note that the trans-eQTLs are defined those that are more than 5Mb away from the probe (this distance threshold can be changed by --cis-wind).
smr --descriptive-trans --beqtl-summary myeqtl --trans-wind 1000 --out mydescriptive
MeCS
MeCS: Meta-analysis of cis-eQTL in Correlated Samples
# Overview
MeCS is a method that only requires summary-level cis-eQTL data to
perform a meta-analysis of cis-eQTLs from multiple cohorts (or tissues)
with sample overlaps. It estimates the proportion of sample overlap from
null SNPs in the cis-regions and meta-analysed the eQTL effects using a
generalized least squares approach. The method can be applied to data
from genetic studies of molecular phenotypes (e.g. DNA methylation and
histone modification).
Bug reports or questions to Jian Yang (jian.yang@westlake.edu.cn) at School of Life Sciences, Westlake University.
# Tutorial
Example
smr --besd-flist my_file.list --mecs --thread-num 5 --out mecs_result
--mecs implements the MeCS analysis.
Specify a p-value threshold to exclude the significant SNPs from calculating the cohort correlation matrix.
osca --besd-flist my_file.list --mecs --pmecs 0.01 --out mymecs
--pmecs reads a p-value threshold to exclude the significant SNPs from calculating the cohort correlation matrix. The default value is 0.01.
Specify a minimum number of null SNPs for calculating the cohort correlation matrix.
osca --besd-flist my_file.list --mecs --nmecs 100 --out mymecs
--nmecs reads a minimum number of null SNPs that are required to calculate the cohort correlation matrix. The default value is 100.
Example
smr --besd-flist my_file.list --meta --thread-num 5 --out meta_result
--meta implements the conventional inverse-variance-weighted meta-analysis assuming all the cohorts are independent.
Citation
Qi T, Wu Y, Zeng J, Zhang F, Xue A, Jiang L, Zhu Z, Kemper K, Yengo L, Zheng Z, eQTLGen Consortium, Marioni RE, Montgomery GW, Deary IJ, Wray NR, Visscher PM, McRae AF & Yang J (2018) Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nature Communications, 9: 2282.
Options Reference
Option
Description
--beqtl-summary
reads summary-level data from a eQTL study in binary format.
--besd-flist
reads a file to get the full paths of the BESD files.
--bfile
reads individual-level SNP genotype data in PLINK binary format.
--bolt-assoc-format
reads eQTL summary data in BOLT_LMM output format.
--cis-wind
defines a window centred around the probe to select cis-eQTLs.
--eqtl-flist
reads a file to get probe information and locations of the eQTL summary data files.
--exclude-exposure-probe
excludes a subset of exposure probes from analysis.
--exclude-outcome-probe
excludes a subset of outcome probes from analysis.
--exclude-probe
excludes a subset of probes from analysis.
--exclude-single-exposure-probe
excludes a single exposure probe from analysis.
--exclude-single-outcome-probe
excludes a single outcome probe from analysis.
--exclude-snp
excludes a subset of SNPs from analysis.
--extract-cis
extracts the cis-eQTLs.
--extract-exposure-probe
extracts a subset of exposure probes for analysis.
--extract-outcome-probe
extracts a subset of outcome probes for analysis.
--extract-probe
extracts a subset of probes for analysis.
--extract-single-exposure-probe
extracts a single exposure probe for analysis.
--extract-single-outcome-probe
extracts a single outcome probe for analysis.
--extract-snp
extracts a subset of SNPs for analysis.
--from-probe
specifies the start probe.
--from-probe-kb
specifies the start physical position of the probes.
--from-snp
specifies the start SNP.
--from-snp-kb
specifies the start physical position of the region.
--gemma-fomat
reads eQTL summary data in GEMMA association output format.
--gene
specifies a single gene to select probes.
--genes
extracts a subset of probes which tag the genes in the list.
--gene-list
specifies a gene annotation file.
--geno-uni
indicates all the input .esd files are identical.
--gwas-summary
reads summary-level data from GWAS in GCTA-COJO format.
--heidi-m
minimum requirement of the number of eQTLs used in the HEIDI test.
--heidi-mtd
specify a method for HEIDI test.
--heidi-off
turns off the HEIDI test.
--keep
includes a subset of individuals in the reference sample for analysis.
--ld-pruning
LD r-squared threshold for pruning SNPs (eQTLs) in HEIDI test, removing SNPs in high LD with the top associated eQTL.
--maf
removes SNPs based on a minor allele frequency (MAF) threshold in the reference sample.
--make-besd
saves summary data in BESD format. By default, the data will be stored in sparse BESD format.
--make-besd-dense
saves summary data of all SNPs for all probes.
--out
specifies filename prefix for output files.
--p-technical
reads a p-value threshold to select technical eQTLs.
--peqtl-heidi
threshold of eQTL p-value to select eQTLs for the HEIDI test.
--peqtl-other
threshold of eQTL p-value to select eQTLs apart from the cis and trans regions.
--peqtl-smr
threshold of eQTL p-value to select the top associated eQTL for the SMR test.
--peqtl-trans
threshold of eQTL p-value for trans-SNPs.
--plink-qassoc-fomat
reads eQTL summary data in PLINK-qassoc format.
--plot
saves in text format the data for plot.
--probe
specifies a single probe.
--probe-chr
specifies a chromosome to select probes.
--probe-wind
defines a window centred on a specified probe.
--qfile
reads eQTL summary data in SMR query output format.
--query
saves in text format a subset of the eQTL summary dataset based on the specified eQTL p-value threshold.
--remove
excludes a subset of individuals in the reference sample from the analysis.
--rm-technical
specifies the probe hybridization region and excludes the technical eQTLs.
--set-wind
defines a window width (Kb) centred around the top associated cis-eQTL to select SNPs in the cis-region.
--smr-multi
turns on set-based SMR test in the cis-region.
--snp
specifies a single SNP.
--snp-chr
specifies a chromosome to select SNPs.
--to-probe
specifies the end probe.
--to-snp
specifies the end SNP.
--snp-wind
defines a window centred on a specified SNP.
--to-probe-kb
specifies the end physical position of the probes.
--to-snp-kb
specifies the end physical position of the region.
--trans
turns on SMR and HEIDI tests in trans regions.
--trans-wind
defines a window centred around the top associated trans-eQTL to select SNPs .
--target-snp
specifies a SNP as the target for the SMR and HEIDI tests as described above.
--thread-num
specifies the number of OpenMP threads for parallel computing.
--update-freq
reads allele frequency file.
Download
Executable Files (version 1.3.1)
smr-1.3.1-linux-x86_64.zip
smr_Mac_v1.03.zip
smr-1.3.1-win-x86_64.zip
The executable files (binary code) are released under MIT license.
Source code
smr_v1.3.1_src.tar.gz
The source code are released under GPL v2.
R script for SMR locus plot
R script and sample file for SMR locus plot:
plot.zip
Update log
35. Version 1.3.1 (20 May, 2022): fixed a few bugs and added a script to compile the source code by CMake.
34. Version 1.03 (25 October, 2019): updated Mecs to allow data management flags.
smr-1.03-linux.zip smr_Win_v1.03.zip
33. Version 1.02 (22 May, 2019): fixed a bug in generating the data file for Omics SMR plot.
32. Version 1.01 (16 April, 2019): updated the log information of MeCS.
31. Version 1.0 (15 January, 2019): 1) Formally released SMR version 1.0; 2) Added flags to query descriptive summary of the cis-eQTL and trans-eQTL data.
30. Version 0.712 (6 September, 2018): Added an option to make and query LD information in binary format (i.e. .bld, and .esi files) and an interface to read LD matrix (in BLD format) as the reference for the HEIDI test.
29. Version 0.711 (30 August, 2018): Added flags --qtltoos-nominal-format and --qtltoos-permu-format to transform eQTL summary statistics in QTLtools output format to SMR BESD format.
28. Version 0.710 (21 June, 2018): improved the allele frequency checking step of SMR (more flexible than the previous version). Note that we also updated the frequencies of the effect alleles in the McRae et al. mQTL data, GTEx eQTL data and Brain-mMeta mQTL data.
27. Version 0.709 (14 June, 2018): added a QC step to check the differences in allele frequency among eQTL, GWAS and LD reference data for the SMR analysis.
26. Version 0.708 (31 May, 2018): added two flags --extract-target-snp-probe and --extract-snp-probe to extract specific SNP-probe pairs for the SMR analysis.
25. Version 0.707 (11 May, 2018): changed the default LD pruning r2 threshold for SMR-multi from 0.9 to 0.1.
24. Version 0.706 (1 April, 2018): fixed a problem of linking a library in Linux and a bug with --target-snp.
23. Version 0.705 (16 Feburary, 2018): Updated MeCS and removed some confusing log information when making BESD files.
22. Version 0.704 (01 Feburary, 2018): Fixed a bug in making BESD files.
21. Version 0.703 (19 January, 2018): 1) Added flags --add-n to add sample size in a BESD file and --show-n to display the sample size on screen or in a log output. 2) Added flag --matrix-eqtl-format to transform the eQTL summary statistics in Matrix eQTL output format to SMR BESD format. 3) Added flag --fastqtl-nominal-format to transform the eQTL summary statistics in FastQTL outformat to SMR BESD format. 4) Added flags --update-epi and --update-esi to update or complete the information in .epi file and .esi file respectively.
20. Version 0.702 (07 January, 2018): Added flags --extract-snp-p and --exclude-snp-p to make a subset of BESD with a p-value threshold.
19. (02 January, 2018): Released the GTEx eQTL summary data in SMR binary (BESD) format.
18. Version 0.701 (22 December, 2017): 1) Updated some parameters used in the HEIDI test. A lower limit of LD r-squared threshold (the default value is 0.05) has been added to remove SNPs that are not in LD or in low LD with the top eQTL. 2) Added a flag --heidi-max-m to specify the maximum number of SNPs used in the HEIDI test.
17. Version 0.69 (7 October, 2017): added features to run
multi-SNP based SMR and SMR analysis of two molecular traits. Also add a
feature to remove technical eQTLs.
16. (12 September, 2017): Luke R. Lloyd-Jones et al. released CAGE
eQTL summary statistics for SMR analysis.
15. Version 0.68 (11 August, 2017): updated the SMR and HEIDI
tests in the trans regions (the previous version focuses only on the top
trans-eQTL locus and the new version will run the tests for all the
trans-eQTL loci one at a time).
14. Version 0.67 (22 June, 2017): updated the functions to make
BESD file by the following strategy: 1) Z* from N(0, 1) given the
p-value. 2) SE* = b / Z* . 3) store b and SE* in BESD. This
adjustment guarantees that the re-computed p-value from b and SE being
exact the same as the original p-value, useful for data with small
sample size.
13. Version 0.66 (10 January, 2017): updated the function to
generate the file for locus plot. The new version is able to read a gene
list with/without strand information.
12. Version 0.65 (12 December, 2016): added a flag (--heidi-mtd)
for users to choose the original approach or a new approach for HEIDI
test.
11. Version 0.64 (8 August, 2016): updated the .esi file format;
updated the HEIDI test (a new method that improves the power of the
HEIDI test); updated the SMR query output format; improved the analysis
to combine multiple BESD files.
10. Version 0.632 (28 June, 2016): added a feature to make a BESD
file from BOLT-LMM output format.
9. Version 0.631 (23 June, 2016): more options to make BESD files
and more memory-efficient when making binary besd files.
8. Version 0.630 (23 May, 2016): updated features to make binary
besd file from plain text file(s).
7. Version 0.628 (11 May, 2016): added a feature to visualize SMR
results.
6. Version 0.620 (12 April, 2016): added a feature to deal with
duplicate IDs.
5. Version 0.619 (4 April, 2016): updated sparse besd format;
updated features to make sparse verison of BESD; added features to query
eQTL summary results; added features to combine BESD files.
4. Version 0.6 (10 Nov, 2015): added features of SMR and HEIDI
test for the trans regions.
3. 12 Oct, 2015: Eigen library and OpenMP were used.
2. 17 Sept, 2015: updated the format of sparse besd file; added a
function to make sparse besd file by extracting information from full
dense besd file; added a function to check quickly how many probes are
associated with a SNP at p < a threshold(e.g. 5e-8).
1. 24 Aug, 2015: first release.
Data Resource
sQTL summary data
# BrainMeta v2 sQTL summary data (n = 2,865)
We developed a method, THISTLE, which uses individual-level genotype and RNA-seq data or summary-level isoform-eQTL data for splicing QTL (sQTL) mapping (Qi et al. 2022). We applied THISTLE, in combination with a complementary sQTL mapping strategy, for sQTL mapping using RNA-seq data of 2,865 brain cortex samples from 2,443 unrelated individuals of European ancestry with genome-wide SNP data. See below for the link to download the full summary statistics of the sQTLs in SMR binary (BESD) format. You can also query or visualize the sQTL summary statistics using the BrainMeta portal.
BrainMeta v2 cis-sQTL summary data (Qi et al. 2022) in SMR binary (BESD) format:
BrainMeta_cis_sqtl_summary.tar.gz (hg19) (9.0 GB)
These are pooled cis-sQTLs identified by THISTLE and LeafCutter & QTLtools. Only SNPs within 2 Mb distance from each gene or intron are available.
BrainMeta v2 trans-sQTL summary data (Qi et al. 2022) in SMR binary (BESD) format:
BrainMeta_trans_sqtl_summary.tar.gz (hg19) (2.3 GB)
These are pooled trans-sQTLs identified by both THISTLE and LeafCutter & QTLtools. Only SNP-gene pairs with distance > 5 Mb or on different chromosomes are available.
eQTL summary data
# BrainMeta v2 eQTL summary data (n = 2,865)
We performed eQTL mapping using RNA-seq data of 2,865 brain cortex samples from 2,443 unrelated individuals of European ancestry with genome-wide SNP data. See below for the link to download the full summary statistics of the eQTLs in SMR BESD format. You can also query or visualize the eQTL summary statistics using the BrainMeta portal.
BrainMeta v2 cis-eQTL summary data (Qi et al. 2022) in SMR binary (BESD) format:
BrainMeta_cis_eqtl_summary.tar.gz (hg19) (2.6 GB).
Only SNPs within 2 Mb distance from each gene are available.
BrainMeta v2 trans-eQTL summary data (Qi et al. 2022) in SMR binary (BESD) format:
BrainMeta_trans_eqtl_summary.tar.gz (hg19) (2.2 GB).
Only SNP-gene pairs with distance > 5 Mb or on different chromosomes are available.
# Westra eQTL summary data
Westra eQTL summary data (n = 3511; Westra et al. 2013 Nat Genet) in SMR
binary (BESD) format:
westra_eqtl_data_hg18.zip (hg18) (10.3 MB)
westra_eqtl_data_hg19.zip (hg19) (10.3 MB)
# CAGE eQTL summary data
CAGE eQTL summary data (n = 2765; Luke R. Lloyd-Jones et al. 2017 AJHG) in SMR binary (BESD) format:
cage_eqtl_data_hg19.tgz (hg19) (3.8 GB)
cage_eqtl_data_lite_hg19.tgz (hg19) Lite version of the CAGE data (only SNPs with P < 1e-5 are included; 86.1 MB)
The CAGE eQTL results have finer coverage than the Westra et al. 2013
results with comparable power. Please note that the EGCUT cohort is
common to both the Westra et al. 2013 and CAGE data sets. Please see the
above link to the CAGE paper that outlines how these eQTL results were
generated.
Please see the associated Shiny App for further interactive interrogation of the results generated in the CAGE analysis.
# V8 release of the GTEx eQTL/sQTL summary data
V8 release of the GTEx eQTL/sQTL summary data (n = 73 - 670; GTEx Consortium 2020 Science) in SMR binary (BESD) format:
GTEx_V8_cis_eqtl_summary(hg19) (48 GB)
GTEx_V8_cis_eqtl_summary_lite (hg19) Lite version of the GTEx V8 eqtl data (only SNPs with P < 1e-5 are included; 506MB)
This is a set of cis-eQTL summary data across 49 human tissues from the GTEx project. Only SNPs within 1Mb of the transcription start site are available. The forth column of the *.epi file is the middle position of the probe sequence rather than the transcription start site.
GTEx_V8_cis_sqtl_summary(hg19) (190 GB)
GTEx_V8_cis_sqtl_summary_lite (hg19) Lite version of the GTEx V8 sqtl data (only SNPs with P < 1e-5 are included; 499MB)
This is a set of cis-sQTL summary data across 49 human tissues from the GTEx project. sQTLs were estimated by testing associations between genetic variants and intron excisions that were estimated from LeafCutter (Li et al. 2018 Nat Genet).
The standard errors in the BESD files were re-computed from the observed effect sizes and p-values based on a chi-squared distribution with 1 degree of freedom. See GTEx Portal for details about the eQTL/sQTL analysis.
# V7 release of the GTEx eQTL summary data
V7 release of the GTEx eQTL summary data (n = 80 - 491; GTEx Consortium 2017 Nature) in SMR binary (BESD) format:
GTEx_V7_cis_eqtl_summary.tar.gz (hg19) (55 GB)
GTEx_V7_cis_eqtl_summary_lite.tar.gz (hg19) Lite version of the GTEx V7 data (only SNPs with P < 1e-5 are included; 5.3 GB)
This is a set of cis-eQTL summary data across 48 human tissues from the GTEx project. Only SNPs within 1Mb of the transcription start site are available. The standard errors in the BESD files were re-computed from the observed effect sizes and p-values based on a chi-squared distribution with 1 degree of freedom. The forth column of the *.epi file is the middle position of the probe sequence rather than the transcription start site. See GTEx Portal for details about the eQTL analysis.
# Qi et al. brain eQTL summary data
GTEx-brain eQTL data (estimated effective n = 233)
GTEx-brain eQTL summary data (Qi et al. 2018 Nat Commun) in SMR binary (BESD) format: GTEx-brain.tar.gz (1.1 GB)
This is a set of eQTL data from a meta-analysis of 10 brain regions in GTEx v6 (GTEx Consortium 2017 Nature) correcting for sample overlap by the MeCS method. Only SNPs within 1Mb distance from each probe are available.
Brain-eMeta (now called BrainMeta v1) eQTL data (estimated effective n = 1194)
BrarinMeta eQTL summary data (Qi et al. 2018 Nat Commun) in SMR binary (BESD) format: BrainMeta_v1.tar.gz (1.1 GB)
This is a set of eQTL data from a meta-analysis of GTEx brain (GTEx Consortium 2017 Nature), CMC (Fromer et al. 2016 Nat Neurosci), and ROSMAP (Ng et al. 2017 Nat Neurosci) by MeCS. Only SNPs within 1Mb distance from each probe are available.
# Geuvadis eQTL summary data
Geuvadis eQTL summary data (Lappalainen et al. 2013 Nature) in SMR binary (BESD) format:
geuvadis_EUR_rsid.tar.gz (hg19) (650.5MB)
Unthresholded Geuvadis eQTL data for lymphoblastoid cell lines isolated from 373 EUR individuals were used, with YRI individuals excluded. The eQTL summary data were from EUR373.gene.K10.noplim.cis_assembled.txt.gz. Since betas are not available for the unthresholded dataset, they were estimated from t-values, allele freqs and n=373 according to Formula (6) in the SMR paper (Zhu et al. 2016 Nat Genet). Geuvadis SNP ids were converted to rsids for compatibility with external GWAS data and plink 1KG files. The data are based on GRCh37 assembly, and gene IDs deprecated in the GRCh37 version of Ensembl were removed. We would like to acknowledge Mikhail Spivakov for his effort in transforming the data to SMR BESD format and writing the description above.
# PsychENCODE eQTL summary data
Here are two sets of cis-eQTL summary data in the prefrontal cortex from the PsychENCODE project (n = 1387). Only the data of SNPs in a 1 Mb window around each gene are available.
a. PsychENCODE eQTL summary data (correcting for 50 PEER factors) in SMR binary (BESD) format:
PsychENCODE_cis_eqtl_PEER50_summary.tar.gz (hg19) (33 MB)
Only SNPs with FDR < 0.05 are available for each gene. The eQTL analyses were performed with 50 probabilistic estimation of expression residuals (PEER) factors included as covariates (see Wang et al. 2018 Science for details about data generation and analysis).
b. PsychENCODE eQTL summary data (correcting for 100 HCP factors) in SMR binary (BESD) format:
PsychENCODE_cis_eqtl_HCP100_summary.tar.gz (hg19) (65 MB)
Similar to the data set above but the eQTL analyses were performed including100 hidden covariate (HCP) factors as covariates (see Gandal et al. 2018 Science for details about data generation and analysis).
mQTL summary data
# Hatton et al. mQTL summary data
Hatton et al. mQTL summary data in SMR BESD format.
EAS.tar.gz (33GB, n=2,099) mQTL summary data from a meta-analysis of samples of East Asian ancestry.
EUR.tar.gz (39GB, n=3,701) mQTL summary data from a meta-analysis of samples of European ancestry.
# McRae et al. mQTL summary data
McRae et al. mQTL summary data (McRae et al. 2018 Sci Rep; Wu et al. 2018 Nat Commun) in SMR binary (BESD) format (n = 1,980):
LBC_BSGS_meta.tar.gz (hg19) (7.5 GB)
LBC_BSGS_meta_lite.tar.gz (hg19) Lite version of the McRae et al. mQTL data (only SNPs with P < 1e-5 are included; 241 MB)
The original mQTL data were generated in two cohorts BSGS (n = 614) and LBC (n = 1366) in peripheral blood (McRae et al. 2018 Sci Rep). The methylation states of all the samples which are of European descent were measured based onIllumina HumanMethylation450 chips. The mQTL summary data available here were a meta-analysis of the BSGS and LBC data (Wu et al. 2018 Nat Commun). Only the DNA methylation probes withat least a cis-mQTL at P < 5e-8 and only SNPs within 2Mb distance from each probe are available.
# Qi et al. brain mQTL summary data
Brain-mMeta mQTL data (estimated effective n = 1160)
Brain-mMeta mQTL summary data (Qi et al. 2018 Nat Commun) in SMR binary (BESD) format: Brain-mMeta.tar.gz (893 MB)
This is a set of mQTL data from a meta-analysis of ROSMAP (Ng et al. 2017 Nat Neurosci), Hannon et al. (Hannon et al. 2016 Nat Neurosci) and Jaffe et al. (Jaffe et al. 2016 Nat Neurosci). In the ROSMAP data, only SNPs within 5Kb of each DNA methylation probe are available. In the Hannon et al. data, only SNPs within 500Kb distance from each probe and with PmQTL < 1.0e-10 are available. In the Jaffe et al. data, only SNPs within 20Kb distance from each probe and with FDR < 0.1 are available.
# Hannon et al. mQTL summary data
Whole blood mQTL data set used in Hannon et al. (2018 AJHG).
Hannon et al. WholeBlood dataset.zip (sample size = 1,175; 121MB)
Three mQTL datasets used in Hannon et al. (2017 AJHG). All the files are in SMR BESD format.
Two blood mQTL datasets (Hannon et al. 2016 Genome Biology)
Hannon et al. Blood dataset1.zip (sample size = 639; 42MB)
Hannon et al. Blood dataset2.zip (sample size = 665; 25MB)
Fetal brain mQTL data (Hannon et al. 2015 Nat Neurosci)
Hannon et al. FetalBrain.zip (sample size = 166; 4.8MB)
Note: 1) The SNPs are coded as chr:bp (based on the genome build hg19)
rather than with rsIDs. 2) SNPs with mQTL p-values > 1e-10 are not included. 3) Any question regarding to these datasets should be addressed to Eilis Hannon or Jonathan Mill.
caQTL summary data
# Bryois et al. chromatin accessibility QTL (caQTL) data
Bryois et al. caQTL summary data in SMR binary (BESD) format:
Bryois_caQTL_summary.tar.gz (208 MB)
This is a set of cis-caQTL summary data from the prefrontal cortex of 135 schizophrenia patients and 137 controls. Only the SNPs in a 50 Kb window around each chromatin accessibility peak are available (see Bryois et al. 2018 Nat Commun for details about data generation and analysis).
什么是SMR技术? | 东芝半导体&存储产品中国官网
什么是SMR技术? | 东芝半导体&存储产品中国官网
Asia-Pacific - 简体中文
Asia-Pacific
English
简体中文
繁體中文
한국어
日本語
Americas
English
Europe (EMEA)
English
Asia-Pacific
English
简体中文
繁體中文
한국어
日本語
Americas
English
Europe (EMEA)
English
联系我们
存储产品首页
网站首页
半导体首页
存储产品首页
公司信息首页
网站首页
查看
半导体
查看
存储产品
查看
产品
选择产品种类
数据中心/企业级存储产品
云级容量
企业级容量
企业级性能
个人存储产品(移动硬盘/内置机械硬盘)
内置/专用存储产品
监控
NAS
PC
视频
最近浏览的产品种类
什么是SMR技术?
关于我们的存储产品
查看
技术中心
查看
支持
支持首页
安全公告
兼容性信息
质保支持
型号含义
停产产品归档
常见问题(FAQ)
购买
查看
公司信息
查看
关键词搜索
型号搜索
交叉搜索
关键词搜索
参数搜索
库存查询与购买
搜索
包含
与开头部分一致
完全一致
搜索
搜索
全部
二极管
MOSFET
双极晶体管
IGBT/IEGT
隔离器/固态继电器
通用逻辑IC
电源管理IC
电机驱动IC
智能功率IC(IPD)
线性IC
微控制器
车载器件
ASSP
无线通信设备IC
传感器
搜索
包含
与开头部分一致
完全一致
搜索
This webpage doesn't work with Internet Explorer. Please use the latest version of Google Chrome, Microsoft Edge, Mozilla Firefox or Safari.
请输入3个以上字符
About information presented in this cross reference
The information presented in this cross reference is based on TOSHIBA's selection criteria and should be treated as a suggestion only. Please carefully review the latest versions of all relevant information on the TOSHIBA products, including without limitation data sheets and validate all operating parameters of the TOSHIBA products to ensure that the suggested TOSHIBA products are truly compatible with your design and application.Please note that this cross reference is based on TOSHIBA's estimate of compatibility with other manufacturers' products, based on other manufacturers' published data, at the time the data was collected.TOSHIBA is not responsible for any incorrect or incomplete information. Information is subject to change at any time without notice.
请输入3个以上字符
This page partially uses JavaScript. This page may not operate normally when these functions are not supported by your browser or the setting is disabled.Please look for the information you need from the following pages:
网站地图
联系我们
Asia-Pacific - English
Asia-Pacific - 简体中文
Asia-Pacific - 繁體中文
Asia-Pacific - 한국어
Asia-Pacific - 日本語
Americas - English
Europe (EMEA) - English
半导体与存储产品首页
存储产品(HDD)
什么是SMR技术?
什么是SMR技术?
说明
SMR(叠瓦式磁记录)会覆写磁道上原本的记录,就像在屋顶上叠瓦片一般。
CMR和SMR各有利弊。
CMR (Conventional Magnetic Recording)传统式磁记录
优点
可以直接将数据重新写入到已写入的扇区
缺点
使用离散磁道会限制磁录密度增加的速度
SMR(Shingled Magnetic Recording)叠瓦式磁记录
优点
与CMR相比,能更有效地利用记录表面积,获得更高的数据容量。
缺点
因为磁道是重叠的,所以不能直接重写至已写入的扇区。 恢复包含过期数据的空间可能会影响性能。
东芝SMR如何帮助保持随机写入性能
东芝创新的媒体缓存设计使硬盘能够针对之前已写入数据集,有效地处理随机写入数据更新。
新数据从DRAM缓冲区临时写入媒体缓存。
在合并旧数据和新数据后,修改后的数据按顺序写入另一个SMR磁段。
这有助于避免在重新写入数据时性能下降,从而改进了先前SMR所具有的缺点。
SMR随机写入方式
此解决方案的成果是什么?
PCMark®是一款行业标准的PC基准测试工具,用于测试PC在系统和元件层面的性能。DT02系列(SMR 5400rpm)比竞争对手A(SMR 7200rpm)的产品拥有更高的性能。
PCMark® Vantage评分
PCMark®元件性能评分
PC环境:
工具:PCMark® Vantage
系统:GIGABYTE H97-GAMING 3 (Intel CoreTM i5-4460 3.20GHz/ 8GBRAM)
驱动器:Intel Desktop/Workstation/Server Express Chipset SATA AHCI Controller v. 12.8.0.1016
操作系统:Windows 10 v1903 x64
产品线
以下产品采用SMR技术。
DT02-VH系列
DT02-V系列
DT02系列
MQ04系列
在新窗口打开
To Search
To Top
网站地图
半导体产品
SiC功率器件
MOSFET
IGBT/IEGT
隔离器/固态继电器
电源管理IC
智能功率IC
线性IC
电机驱动IC
二极管
双极晶体管
微控制器
车载器件
应用于无线通信设备的IC
通用逻辑IC
移动外围设备的桥接IC
射频器件
传感器
线性图像传感器
存储产品
数据中心/企业级存储产品
内置/专用存储产品
个人存储产品(移动硬盘/内置机械硬盘)
电子杂志
订阅电子杂志
公司信息
SNS
东芝微博二维码名称:东芝半导体
扫一扫关注我们
×
东芝微信二维码名称:东芝半导体
扫一扫关注我们
×
English
简体中文
繁體中文
한국어
日本語
English
English
English
简体中文
繁體中文
한국어
日本語
English
English
隐私政策
网站使用条款与条件
Cookie设定
联系我们
沪ICP备19048049号-1
Copyright © 2024 TOSHIBA ELECTRONIC DEVICES & STORAGE CORPORATION, All Rights Reserved.
沪公网安备 31010602002607号
关于机械硬盘的 CMR 和 SMR 。不买叠瓦盘!!! - 知乎
关于机械硬盘的 CMR 和 SMR 。不买叠瓦盘!!! - 知乎切换模式写文章登录/注册关于机械硬盘的 CMR 和 SMR 。不买叠瓦盘!!!长斋慎独通信工程CMR和SMR是机械硬盘的两种不同数据记录方式。CMR翻译过来的中文意思是传统磁记录方式,采用该技术的硬盘会在磁道间保有保护间距,数据不会被重复叠写,安全稳定性更高。这里再延申一下,其实在更早之前还有一种叫PMR的垂直磁道记录技术,后来随着技术的发展,PMR细分出了CMR和SMR,一般我们认为PMR=CMR。一、CMRCMR 传统磁记录方式二、SMRSMR被称为叠瓦式磁记录方式,顾名思义该技术是把硬盘中的磁道像瓦片一样重叠堆放在一起,它的优点是提升硬盘的存储密度,从而增加约25%的容量。但是SMR也有缺点,就是它不适合频繁改写(数据),由于其磁头读头小、写头大,叠瓦磁道密度高的原因,会影响磁盘的写头工作,在随机写入或改写内容时,会把相邻磁道也一起改掉,因此叠瓦通常叠几圈以后会有一行正常宽度的磁道,把磁盘分成一个个区域。要改写内容,需要把这个数据所在的整个区域的内容先转存到缓存里,在缓存里改好,再一起写回去。由于SMR硬盘并不适合频繁读写,,我们应尽量避开SMR硬盘。适合备份。三、如何区别部分硬盘品牌商不会主动标记CMR和SMR,可能1-4TB的款式会用CMR技术,4-12TB则用SMR技术,从而出现CMR和SMR混用的情况,买硬盘犹如抽奖。当然,我们大可以直接选购选购像标明了“CMR PMR”技术的产品,希捷酷狼系列NAS硬盘(NAS硬盘是智商税)官网查询型号 确认使用技术 !!希捷:西数:HDD硬盘行业最近两周遭遇“SMR门”,西数、希捷、东芝三大厂商都陷入了这一波的争议中,不过这三家厂商态度还不错,陆续开始明确标明SMR硬盘与CMR硬盘的型号,西数今天已经在官网规格表上全面更新。对于SMR与CMR硬盘,我们之前专门介绍过SMR技术的优点与缺点,简单来说就是它可以让HDD硬盘在没有技术革命的情况下就提升25%的容量,但代价就是写入性能、可靠性降低,这需要厂商开发更好的SMR管理技术来弥补。对于SMR硬盘,三大厂商放弃是不可能放弃的,SMR硬盘的占比只会越来越大,现在大家需要的是厂商将SMR硬盘与CMR硬盘明确区分出来,而不像是之前那样混在一起,至少给消费者选择的权利。随后希捷、西数、东芝都公布了SMR硬盘的具体型号。对西数来说,3.5寸蓝盘中的2TB(WD20EZAZ)、6TB(WD60EZAZ)为SMR、2.5寸蓝盘中的1TB(WD10SPZX)、2TB(WD20SPZX)是SMR,2.5寸黑盘中的1TB(WD10SPSX)也是SMR。4月30日开始,西数的官网规格表也升级了,目前可以详细查看旗下硬盘是否为SMR硬盘。西数官网上蓝盘型号中SMR硬盘编号西数官网上红盘型号中SMR硬盘编号西数官网上黑盘型号中SMR硬盘编号发布于 2021-03-02 11:37硬盘SMR(叠瓦式磁记录)网络附加存储(NAS)赞同 29247 条评论分享喜欢收藏申请
如何判定一块机械盘是不是采用叠瓦式磁记录(SMR)? - 知乎
如何判定一块机械盘是不是采用叠瓦式磁记录(SMR)? - 知乎首页知乎知学堂发现等你来答切换模式登录/注册笔记本硬盘机械硬盘如何判定一块机械盘是不是采用叠瓦式磁记录(SMR)?如题,现在选购机械盘想尽可能避免踩雷。毕竟SMR还是太坑了。 以及,现有哪些型号的在产硬盘仍然采用CMR技术?显示全部 关注者242被浏览570,125关注问题写回答邀请回答好问题 25添加评论分享9 个回答默认排序知乎用户更新!西数已经公布出所有使用SMR技术硬盘的列表,并且未来也会更新,参见如下链接:希捷也公布了使用SMR技术硬盘的列表,链接如下:如何判断一块硬盘是否为SMR?官方已经在一些盘的数据表里注明了CMR,但一些老型号和真正的SMR盘却没有写。得到官方确认自然最好,但咱们还是要更进一步去了解真相。希捷酷鹰的数据表中注明了记录技术为CMR首先看缓存是不靠谱的,因为很多大容量硬盘用的是256M缓存的CMR,有的使用64M缓存却也是SMR,比如西数2.5寸1T黑盘WD10SPSX,它只有64M缓存,之前大家一致认为这不是SMR,结果被西数打脸打得啪啪响。容量/碟片数=单碟容量,这个计算不算很靠谱,但在没有更详细资料的情况下可以判断,它分为2.5寸和3.5寸。3.5寸硬盘,计算出的单碟容量在1.7T以内是CMR,大于1.7T,或者单碟容量大于等于2T的,可以肯定是SMR。但这个方法也不一定靠谱,因为厂商可能会节约成本或者其他原因,将小于1.7T也搞成SMR,证据见下图。接下来告诉大家一个新的方法来判断一块硬盘是否为SMR。我们要上硬盘厂商的官网,搜索硬盘的型号,然后找到硬盘的规格参数。拿希捷的监控盘ST6000VX001举栗子,看看它到底是不是CMR盘。官网搜到硬盘型号后,打开硬盘系列的介绍页,这里点击规格。按ctrl+F,查找你要找的型号,点击蓝色的图标——用户手册,打开pdf文档。注意型号必须完全对上在打开的pdf文档中找到“Drive Specifications”,然后关注“Track density, KTPI (ktracks/in avg.)”、“Areal density (avg)”这两组数据,这两个参数分别是轨道密度和面积密度。轨道密度越大,磁道间距越小,小到一定程度,受限于当前的磁头技术,轨道宽度比写磁头还要窄,磁头写入时会破坏临近磁道数据,这就是叠瓦盘了,因此它的轨道密度才会如此之大。但是不可能将所有磁道全部重叠,不然中间修改一点点就会导致全盘重写了,必须留有CMR缓冲区以供随机写入,这个CMR缓冲区也是有大有小,这也是叠瓦盘的密度不一样的原因之一。CMR缓冲区就像TLC的SLC缓存一样,没写满时,速度是很快的,一旦写满,速度就会直线下降。但SMR盘不像TLC,不允许直接写入SMR区域,因此这个缓冲区一旦写满,就会触发硬盘内部整理,和SSD不同的是,机械硬盘只有一组磁头,整理时难免顾此失彼,也就是100%占用、读写卡顿的来源。这是希捷银河X16,也是目前CMR容量的极限,轨道密度423,面积密度1028这是希捷酷鱼ST2000DM008的参数,轨道密度为540,实锤SMR盘这是希捷官网查到的酷鹰部分型号的参数,ST6000VX001也在其中,它的轨道密度是540,和上面的ST2000DM008一样,属于SMR。其中3T和6T计算的单碟容量都是1.5T,表面密度为900,又不属于SMR,猜测是因为两者所用的工艺不一样,厂商为了节省成本,采用了一样的盘体和工艺,根据不同容量装入不同数量的盘片希捷酷鹰ST6000VX001这块盘其实是有争议的。根据官方公布的SMR列表,它并不是SMR盘。但从轨道密度来判断,它又是实打实的SMR盘,不过它的面积密度却比CMR极限的X16还要低,只有900Gb/in²,X16则是1028Gb/in²。上面提到,SMR技术是因为磁轨宽度比写磁头还要窄,写入时会覆盖临近磁道。厂商出于成本等原因考虑,8T和6T都采用了和SMR相同的盘体,盘片的磁道划分是在硬盘生产时就确定的,但通过固件控制,可以把临近的磁道留空,不去写入,隔一条磁道再去写入,就不会出现覆盖临近磁道数据的问题了,自然这块6T也就不一定是SMR盘了。而它旁边的8T盘,它的轨道密度跟ST2000DM008差不多,肯定是SMR盘无疑了。同理ST3000VX009是CMR盘,它旁边的4T和2T都是SMR盘。下面是ST6000VX001和ST3000VX009在2020年7月1日新修订的文档,其中去除了2T、4T和8T的盘,以及在后面2.4小节标明了硬盘所用的技术,希捷的多个文档都已更新这个部分,以后可以直接看这里来确认硬盘所用的技术了。不过部分文档并没有这个,还是要根据前面的方法判断。既然尘埃落定,这块6T的监控盘确认是CMR,大家可以放心购买。这是希捷新修订的文档,其中只包含了ST6000VX001和ST3000VX009,它们的轨道密度都是540,面积密度是900900Gb/in²ST6000VX001和ST3000VX009都已经标明所用的是CMR,大家可以放心购买丧心病狂的希捷连500G笔记本硬盘都不放过……这种笔记本硬盘的轨道密度比台盘还高,也就意味着CMR缓冲区更小,数据整理更频繁,寿命也就更差。这种盘多了一种暴毙的原因,磁头“累”死了顺便提一下监控盘为什么也可以做成SMR。我们的监控机通常都是顺序写入数据的,不同系统可能算法不一样,写入的方式也不一样,但大体上还是顺序写入的,一般是按日期、时间生成视频文件。如果硬盘(阵列)写满了,要么报警要求手动清理早期的视频,要么会直接删除最早期的视频。删除文件一般是将文件从文件系统中删除索引,留在原地的数据就会被新的视频数据覆盖掉。如果我们有TRIM技术,告知硬盘哪些数据可擦除,普通的SMR盘可能就会去搬迁整理了,但为监控定制的固件可能不会,它只会记录哪些band可以直接擦掉,哪些需要做一些小迁移,或者干脆直接丢弃整个band,也就是256MB数据也没什么大不了的,毕竟实时写入重要。当然前提是硬盘必须知道自己处于监控盘模式,不然消费者买来当普通盘用可就危险了。因为有了上面的这种处理方式,监控级硬盘处于监控模式时几乎不会出现100%占用的问题,这样也就避免了阵列因为硬盘不响应而将其踢出阵列的问题。我们通常所知的SMR为DMSMR,也就是硬盘固件管理的SMR,性能下降不可预测。但其实还有HMSMR(主机管理的SMR)和HASMR(主机感知的SMR)。其中HMSMR不是咱们民用的,操作系统基本不支持。它需要定制的文件系统和操作系统去主动管理硬盘内的SMR数据。当然监控机也可以定制这样的系统,直接去管理SMR数据,根本不需要STL转换层,性能自然要比DMSMR好得多。3.5寸盘小于等于1TB的盘不用考虑是不是SMR,PMR单碟就能达到的容量,厂商不会吃力不讨好用SMR。2.5寸9㎜的1TB盘也不用担心,最好买HGST 7200转或西数的黑盘,性能更好。倒是2.5寸7㎜的1TB及以上的盘。就很有可能是SMR盘了。2.5寸500GB已经有实锤的SMR盘了。为了省成本,盘体都是一样的,只是格式化出来的容量不同。第一,看缓存。SMR的特性是写入一小块,挪移一大堆。所以必须要大缓存来干这事,才不至于速度非常慢,所以128MB是起步,希捷都用上256MB了。不过大缓存的致命缺点是,一旦断电,缓存的数据来不及保存,造成数据丢失的损失更大,而且SMR还有各种大挪移,损失更加惨重。不过光看缓存不一定准,一些大容量PMR也用上了土豪级的大缓存。第二,去官网查对应型号的碟片数,一般都是在技术参数的文档里,根据总体容量计算单碟容量。现有的PMR技术,在2.5寸一般是500G左右,最大可以到700G,在3.5寸一般是1TB左右,最大1.5TB,单碟容量一旦超过这个值,就大概率是SMR盘了。单碟容量1.5T已经有实锤的SMR盘了,原因同上,省成本第三,装个Crystal Disk Info,查看硬盘支持的技术,如果发现机械硬盘支持TRIM,那就100%是SMR盘了,因为SMR盘的原理,会出现写入放大,TRIM可以让硬盘在空闲时主动清理(覆盖)不需要的数据(垃圾回收),而不是头铁一股脑全部挪移,可以一定程度提升性能,现在新出的SMR盘很多都带这个技术。而PMR本身就是随写随擦的,根本不需要TRIM,使用TRIM甚至会在一定程度上降低性能。第四,避开SMR的热门型号。可以选择监控盘代替消费级硬盘,监控盘由于为大量写入场景设计,因此一般不会使用SMR技术,不然一旦内部进行整理,会导致一部分录像无法正常写入而丢失,同时SMR对阵列也非常不友好,一旦长时间无响应,就有可能被踢下线。不过监控级硬盘的转速一般比较低,对读写速度要求较高的场景不推荐。编辑于 2020-09-04 17:46赞同 414136 条评论分享收藏喜欢收起知空白 关注比较简单但不完全准确的判断方法是看缓存大小(针对2T以上容量硬盘)。(事实证明,其实准确率很难讲高不高)市售常见256兆缓存的较多是SMR,128兆的一部分是SMR,64兆缓存的一般都是CMR。缓存再小那就是很多年前的货了,那会儿没开始大规模应用SMR技术。也不一定完全准确,如果想要准确的判断,请阅读官方手册。因为还是有很多型号不太符合这个判断方法。这个答案我现在第二次修改,因为觉得按照原来说的上面的那个看缓存来判断的方法还是有较大误差。具体分析如下:1.最底部分享的链接里是CHIPHELL网友整理的3.5寸硬盘详细资料,我看了一遍,发现其中一共105个型号的硬盘缓存达到256MB或者512MB。这105个型号中,有15个型号使用SMR技术,90个型号使用CMR技术。这样来说,大缓存硬盘使用SMR技术的型号比例远低于CMR。但我又观察了一下,发现这15个SMR型号基本上都是希捷的盘,而且是同等容量中较为常见的盘。参考希捷在零售市场的占有率和装机市场的畅销程度,这些SMR盘的出镜率是较高的。所以很可能会给消费者留下“大缓存硬盘使用了SMR技术”的印象。其余90个大缓存CMR盘很可能因为在消费市场出镜率低一些,没有给人留下更广泛的印象。所以说,单纯用缓存大小来判断硬盘是否使用SMR技术,不仅仅是不完全准确,而应该是非常不准确了。最后,从chiphell和pceva搬运一些其他大拿的资料给大家Seagate的Archive v1和v2虽然缓存128M但却是SMR;充氦气的X12缓存256M反而是CMR。(pceva版主nighttob提供)西数红盘WD80EFAX以及对应的日立第三代HE10,虽然是256M缓存,但是用的是cmr技术。(pceva网友sss668800提供)CHIPHELL大拿整理的几乎所有市面能见到的3.5寸HDD详细资料,非常强大:链接: https://pan.baidu.com/s/1ickp1VYmOJ-4rx7rhd2o9w 提取码: smuy 如果原有答案给知友造成困惑,在此道歉!编辑于 2020-04-20 12:03赞同 8624 条评论分享收藏喜欢
西数、希捷、东芝全系列概览 + SMR避坑指南 - 知乎
西数、希捷、东芝全系列概览 + SMR避坑指南 - 知乎首发于生活研究所切换模式写文章登录/注册西数、希捷、东芝全系列概览 + SMR避坑指南什么值得买已认证账号SMR避坑指南希捷,2014年首家推出一尖端技术:全球首款 SMR 硬盘,这会将第一代产品中的面密度提高 25%。我们来看看其是如何宣传和科普的↓官网截图东芝篇↓东芝近期公布了《关于东芝消费级存储产品中采用SMR技术的硬盘型号》,文中详细列出了旗下使用SMR技术的产品。东芝官网截图目前东芝P300的1-3TB较为安全,3TB版本也是性价比首选↓与P300同定位的DT01系列1-3T也为垂直硬盘,3TB版本依然是性价比首选,跟P300哪个便宜买哪个↓↓同时东芝官网产品介绍页面也非常详细的列出了其系列产品使用了那种技术;官网截图西数篇↓西部数据官网同样非常详细的列出了旗下使用SMR技术的产品和型号,我已帮值友们整理好,值友们可直接移步全系列导购图查看,也可直接访问西数官网查看;部分型号CMR/SMR一览蓝盘3TB、4TB性价比较高,且都垂直硬盘;↓蓝盘3TB版本适合预算低的消费者,性价比比4T版本稍低;↓紫盘相比蓝盘,提供了三年质保,其他参数基本一致,性价比更高;↓红盘Pro全系都采用垂直技术,可放心选购,最高容量为14TB,就是贵了些;↓希捷篇暂时未能搜集到相关信息,后期若搜集到相关信息,会在文末进行补充。在JD自营平台,酷狼品牌产品明确标明其为垂直硬盘,可放些购买,最高容量为16TB;↓选购要点↓之前写过几篇机械硬盘选购指南,可供值友们参考; 这里简单总结一下:需求确认若是小白,就按需求选购,如装机用就在PC类别中选择,监控用就选在监控类别中选择;如果老手(大佬请带带我), 可在类别选择上适当偏移,如NAS用可以选购企业级,当下载盘用可以在监控盘中选择(如果非要扯监控盘不能家用的话,当下载盘应该问题不大,下载软件如Tr会做数据校验的);叠瓦or垂直注意区分垂直和叠瓦;质保质保年限:2年、3年、5年;质保方式:质保、换新;容价比 = 价格 / 容量,表示每TB容量的单价,可作为选购参考;西数希捷东芝全系列导购图概览西数希捷东芝全系列导购图(长图预警!!!)导购图问答导购图中数据从何处而来?↓导购图中数据均摘自各自品牌的官网,手动录入,可能存在误差,请值友们在留言中指正,方便笔者后期修改;为什么导购图中,三个品牌排版不一样?↓数据来源不一样,且时间跨度较长,前后个人思维方式不同导致,值友们喜欢什么样子的排版可以给我留言,方便笔者后期修改;为什么导购图中,希捷品牌产品未标注CMR/SMR?↓暂时未收集到希捷的相关信息,后期若搜集到相关信息,将会在文末进行补充;为什么导购图中,没有目前热门的东芝P300等产品数据?↓暂未能在东芝官网搜集到相关信息,后期若搜集到相关信息,将会在文末进行补充;目前在售的机械硬盘最大容量是多少?目前在售最大容量为16TB,年内有更大容量的机械硬盘上市,敬请期待。by:什么值得买电脑外设领域作者厕所墙上挂国画本内容来源于@什么值得买http://SMZDM.COM原文链接:一站式解决机械硬盘选购难题 篇一:西数、希捷、东芝全系列概览 + SMR避坑指南发布于 2020-06-16 17:59威腾电子/西部数据(Western Digital)机械硬盘SMR(叠瓦式磁记录)赞同 42988 条评论分享喜欢收藏申请转载文章被以下专栏收录生活研究所让每一次消费产生
机械硬盘(HDD),详解LMR和PMR,CMR和SMR的区别和选择 - 知乎
机械硬盘(HDD),详解LMR和PMR,CMR和SMR的区别和选择 - 知乎切换模式写文章登录/注册机械硬盘(HDD),详解LMR和PMR,CMR和SMR的区别和选择鹿小明一、机械硬盘机械硬盘(来源:wikipedia.org)机械硬盘(Hard Disk Drive,HDD)一般指温彻斯特硬盘,以磁信号存储信息的一种非易失性存储器,也可称为传统普通硬盘或磁盘。组成:盘片,磁头,盘片转轴及控制电机,磁头控制器,数据转换器,接口,缓存等几个部分。工作原理:盘片(磁盘)在电机驱动下高速旋转,悬浮在盘片上方并能沿盘片的直径方向移动的磁头,通过电磁流读写盘片磁道上的信息。这是一个磁盘面的简略俯视图简略盘面俯视图摇臂:带动磁头,摇臂另一端连接主电机;磁头:用于读写磁性记录颗粒的信息,分为读取磁头,写入磁头,写磁头比读磁头大;磁道(Track):经过磁化处理的若干同心环,是实心环,是有横截面宽度的;磁道间有间隙,以保证磁头在各个磁道上读写数据时,不会干扰相邻磁道。磁头在盘面磁道读写数据:盘面侧视图:(来源:wikipedia.org)二、LMR 和 PMR,CMR 和 SMR 概念机械硬盘以磁信号记录数据,这几个简称就是描述机械硬盘所采用的磁记录技术。LMR:水平磁记录(Longitudinal Magnetic Recording)PMR:垂直磁记录(Perpendicular Magnetic Recording)CMR:传统磁记录(Conventional Magnetic Recording)SMR:叠瓦磁记录(Shingled Magnetic Recording)他们的关系如下图:关系图2.1 LMR 和 PMR 概念LMR 和 PMR(来源:wikipedia.org)LMR 水平磁记录:技术最早期,磁性记录颗粒的磁化方向是平行于盘面的;随着颗粒密度提升,端对端磁极互斥会导致一系列问题,失去稳定性,所以 PMR 垂直磁记录技术逐步淘汰了 LMR 技术。PMR 垂直磁记录:磁性记录颗粒的磁化方向是垂直于盘面的。2.2 CMR 和 SMR 概念在 SMR 出现之前,PMR 垂直磁记录就只有一种方式,PMR 的概念很单一,不会混淆。原先的 PMR 再想在磁道上提升磁性记录颗粒很难了,因为磁头和磁性颗粒都很难再小了。所以 SMR 在原先 PMR 的基础上,改变了读写磁道的宽度,以此提升了记录密度。SMR 叠瓦磁记录:相比最初的 PMR 垂直磁记录技术,相当于提升了磁道密度,但核心没变,还是垂直记录方式。CMR 传统磁记录:为了和 SMR 区分,原先的 PMR 被称为 CMR 传统磁记录。PMR:现在的 PMR 概念包含了 CMR 和 SMR 两种垂直记录方式。从消费端来说,市面上的机械硬盘只有 CMR 和 SMR 两种,如果商品宣传页只标 PMR,那就是在含糊其词,想忽悠人。可以去看官方硬盘 datasheet 栏,标注也是 CMR 或 SMR。三、 SMR 和 CMR 磁道区别?SMR 和 CMR 都属于 PMR 垂直磁记录,SMR 磁性记录颗粒的磁化方向没变,还是垂直于盘面的,那一定是在其他地方动了手脚。SMR 叠瓦磁记录相当于把 CMR 盘面上的磁道变窄,从而进一步提高了记录密度。网络上大多数对 SMR 的描述是,像盖房子叠瓦片一样把磁道叠到一起,有点费解。个人认为 SMR 从理解层面上来说,相当于把原来的宽磁道变成了窄磁道,以此提高了记录密度。而宽磁道变窄磁道,当尺寸更大的写入磁头写入数据时,会影响相邻磁道,为了保证相邻数据的正确性,就要频繁读写以缓存相邻的数据,导致了有效读写性能差。3.1 CMR 传统磁道磁头是有大小的,所以磁道之间要给磁头预留空间,各个磁道之间存在空隙,同时写磁头比读磁头大,间隙也是以大的写磁头为标准预留。这样磁道宽度够大,读磁头或写磁头经过某一磁道,都不会干扰到其他磁道,开篇图就是 CMR 传统磁记录的样子。3.2 SMR 叠瓦磁道为了提高密度,SMR 充分利用了磁道之间的空隙和读写磁头的相对大小,以较小的读取磁头为标准,来预留磁道宽度和间隙。只读取数据,问题不大,读取磁头经过某一磁道,和其他磁道没有干扰。但是写入数据就麻烦了,写入磁头更大,磁道宽度小,经过某一磁道,必定影响下一个磁道。如图所示,绿色代表写入磁头,在磁道 #1 写入黄色 New Data 时,影响到了磁道 #2,写磁头经过一次,但影响两个磁道,如果不加以管理,磁道 #2 同样会写入 New Data。叠瓦磁记录示意(来源:wikipedia.org)所以数据写入目标磁道(图例为 Track #1)前,会先标记受影响的磁道(图例为 Track #2),把受到影响的数据读写到别的地方(机械硬盘缓冲区),等目标磁道写入完成,再把数据读写回来。这也就是为什么 SMR 机械硬盘缓冲区要比 CMR 缓冲区大。一次写操作,来来回回要搞三趟。SMR 相对 CMR 做的无用功太多,缓冲区大也无济于事。所以 SMR 写入性能差。刚才说只读取数据,问题不大,确实,数据先写入一次,之后就不改了只读取,但这样的使用场景在日常中太少了,除非是当作纯粹的仓库盘。其实纯读取数据,由于磁道过近,性能也会受到一些影响。日常使用一定会涉及到读写,一旦开始写,读磁头就会去帮写磁头,读取有效数据的能力会大大降低。所以 SMR 在使用体验上相对比 CMR 来说,就是读写能力都差。图示中 SMR 磁道密度和原来比似乎直接翻倍了,实际并没有。使用 SMR 技术的磁盘总体容量比普通磁盘的高出 25%(来源:http://wikipedia.org)四、SMR 和 CMR 选择从消费端来说,市面上的机械硬盘只有 CMR 和 SMR 两种,都属于 PMR 垂直磁记录。如果商品宣传页只标 PMR,那就是在含糊其词,想忽悠人。SMR 提升的数据容量,并不能弥补性能上的劣势,换句话说,牺牲那么大的性能,就提升了一点数据容量,完全不够看。关键点暴击是 SMR 相对于 CMR 也没有价格上的优势。再说一句厂商刚推出 SMR 的时候压根不在宣传端区分,直接混着卖,现在标的清楚是因为事情败露被消费者暴捶!估计这也是部分人抵制 SMR 的原因。一句话总结:选择机械硬盘,认准 CMR 字样!!!发布于 2023-06-10 02:14・IP 属地山东机械硬盘硬盘CMR赞同 121 条评论分享喜欢收藏申请
为什么目前市面上的机械硬盘大都不标注是否使用SMR技术? - 知乎
为什么目前市面上的机械硬盘大都不标注是否使用SMR技术? - 知乎首页知乎知学堂发现等你来答切换模式登录/注册科技笔记本电脑硬盘机械硬盘数据存储(广义)为什么目前市面上的机械硬盘大都不标注是否使用SMR技术?据题主所知,SMR硬盘在改写数据时会覆盖掉邻近磁轨的数据,所以需要多次回写,导致 掉速、可靠性下降、发热、噪音等问题。如果题主没理解错,使用SMR技术…显示全部 关注者575被浏览335,188关注问题写回答邀请回答好问题 132 条评论分享12 个回答默认排序知乎用户======20190907更新======现在越来越多的SMR盘开始支持trim了,其实支持trim的话,比起性能提升,说不定更糟。之前没有trim的时候,一般是PMR缓存写满了才触发搬迁,或者闲置时硬盘内部处理,偷偷搬完了,而PMR缓存一般会比较大,刚开始用不会有太大影响,反而是用着用着,可用空间不足了,体验才会大幅下降。而trim指令是操作系统发出的,操作系统一般会在空闲时发出trim指令,硬盘接收到trim就会立即不分青红皂白地搬迁,这样的话,硬盘性能就有很大的随机性了,很可能跟着SSD一起trim,但SSD的trim多快啊,要知道SSD可以同时写入多个颗粒,硬盘只有一组磁头,不可能同时写多张盘片,所以SMR盘就很可能就被SSD连累导致100%占用性能下降了。本人做过实验,硬盘是ST2000DM008(支持trim),在用USB硬盘盒连接这块盘,往里面拷东西,基本拷到一半时开始100%占用,等待占用结束后接着拷,最终将盘拷满,总共只出现过2次100%占用,但持续时间都不长,最终2t拷满和PMR盘拷满的用时相比会长一些。如果将这块盘用SATA线连接放入机内,再进行如上拷贝操作,会发现它会频繁地出现100%占用,尤其是你只是在拷东西,没干别的操作的时候。大概是操作系统认为系统目前空闲,就发送了trim指令给所有的盘。不支持trim的自然就略过了,支持trim的盘就会开始干活,SMR就此遭殃。最终由于卡顿严重,耗时过长,不得不中止实验。这个实验和之前用的两块2t SMR笔记本盘的结论是类似的,这两块盘都不支持trim。当初发现硬盘严重掉速的时候,基本上2t都快满了,100%占用不算频繁,多是出现在下载中。而后来买了酷玩的混合盘,盘体仍然是SMR。在将文件拷过来的过程中,也是很顺利的,100%占用就出现过一两次。同样的结论也出现在PS4备份这个过程中。还是ST2000DM008(支持trim),用USB硬盘盒连PS4,使用PS4备份,整个过程中应该是完全没有出现过100%占用的情况,速度很稳定。总共800GB的数据4个小时就备份完了,然后PS4换固态后恢复数据的用时基本一致。那么为什么支持了trim反倒出现这种问题了呢?其实这应该是固件对trim指令的处理有关。固态盘在负载低或空闲时,固件会对数据进行搬动整理操作,trim指令实际上就是主动告诉SSD去做这个整理操作。由于固态只能先擦除再写入,为了提升性能,进行垃圾回收操作天经地义,所以在整理时,就要先把把有用的数据搬出来,再清空垃圾所在的块。SMR的操作也是类似的,同样是搬出有用的数据,再清空垃圾所在的区域,这无疑就会造成写入放大。由于机械硬盘性能限制,这种主动回收的策略就有可能造成长时间的占用,导致体验下降。============本人其实是不反对SMR技术的,毕竟在提升单碟容量的同时,减少了盘片数量,同时也减少了盘片、磁头的故障率,当然也有售价。作为仓库盘,SMR还是不错的选择。手里的西数3.5寸移动硬盘一个6TB、一个8TB的盘都基本确定是SMR了,日常仓储使用基本没啥问题。刚才看了下,西数10TB充氦盘2599的价格真香。想当年买个双6TB的移动盘就接近5000块了。但是,请厂商不要把1TB(7㎜笔记本盘)2TB、3TB(台盘)的日常用盘也做成SMR好吗?现在希捷2TB 7200转只剩下SMR盘了,PMR盘就是监控、NAS这类5900降速盘了,想要7200转?上企业级吧。西数也不例外,蓝盘2TB是SMR,4TB居然还是PMR什么鬼。红盘紫盘还没有沦陷,转速一贯的5400,黑盘7200转,性能强劲,价格感人。现在市场上仅存的3.5寸2TB 7200转硬盘就只有东芝P300了,一定要守住啊!===20190330更新===ST2000DM008这个刚出来就大火然后被喷成筛子的单碟2TB硬盘,现在也终于支持TRIM了,这究竟是好事还是坏事呢?固件能不能更新,让原先不支持TRIM的盘支持呢?支持了TRIM之后,对掉速还有磁盘性能的提升能有多大呢?其实不要高看TRIM在SMR盘上的性能,虽然TRIM垃圾回收将一部分空间腾出来而避免数据的写入放大,也可以提升写入性能,但它可能带来的副作用是,发生100%占用的概率变大,但单次持续时间变短,可以理解成将一次长时间的整理分散到整个使用时间里,具体调度由操作系统控制,也是不可预知的,一旦操作系统向硬盘发送TRIM指令,硬盘就会开始整理数据,这时候就会出现100%占用了,而不会等到缓冲区满了固件才去整理。ST2000DM008支持TRIM===20190220更新===惊闻某捷的SMR盘支持trim了?查了下,真的有……不支持Trim的SMR盘支持Trim的SMR盘这两个盘是同型号的,我也有一块这个型号的盘,当时的固件是不支持Trim的。Trim是什么鬼呢?摘自百度百科:TRIM是操作系统告诉NAND闪存固态存储设备要擦除哪些数据的接口指令。当相关页面的数据可以被覆盖时,操作系统会发给SSD一个TRIM指令。SSD控制器等到主机开始删除和再次写入操作的时候,执行安全擦除操作。因为在写入操作过程中不用花时间去擦除原本的数据,写入速度要快得多。当我们在操作系统中删除一个文件时,系统并没有真正删掉这个文件的数据,它只是把这些数据占用的地址标记为‘空’,即可以覆盖使用。但这只是在文件系统层面的操作,硬盘本身并不知道那些地址的数据已经‘无效’,除非系统通知它要在这些地址写入新的数据。在传统的HDD上本无任何问题,因为HDD允许覆盖写入,这也是很多数据恢复软件的工作原理。只要被误删的数据没有被覆盖,数据恢复软件就可以将它们找回。但到SSD上问题就来了,由于NAND修改数据需要先擦除再写入,在没有Trim的情况下,SSD无法事先知道那些被‘删除’的数据页已经是‘无效’的,必须到系统要求在相同的地方写入数据时才知道那些数据可以被擦除,这样的话势必会造成写入性能下降。而用上了TRIM,SSD固件在空闲时将已删除、没有用的数据进行擦除(GC垃圾回收),为将来新数据的写入打基础。那么在SMR盘上用TRIM有什么卵用呢?前面提到,SMR盘修改数据,势必会影响周围的数据,因此SMR盘修改数据时,需要读取一大片后续的数据,并将它们和新数据一并写入。久而久之,整个盘上的数据就会非常混乱,而其中有很大一部分是已删除的数据,但固件并不知道这部分数据是否还有用,在写入新数据时,还在老老实实地进行搬迁,除非系统要求覆盖,固件才会知道这部分数据已作废,可以覆写。覆写的过程中,也不可避免触发数据搬迁的操作。用上了TRIM之后,系统会及时地通知固件哪些数据不需要可以清理,固件也会及时地进行GC垃圾回收操作,可以在一定程度上提高性能,也有可能会在GC时有读写操作而出现性能下降,当然代价就跟SSD一样,失去了误删数据恢复的机会。===20181208更新===发现一篇老外写的SMR的论文,链接粘贴出来如下:===20181206更新===现在发现,越来越多的厂商开始用SMR盘了啊,这里再挂一个吧。这位朋友用的笔记本电脑,出厂自带一块东芝的1T盘,型号为MQ02ABF100,查了下资料发现,这是7mm的盘,单碟1TB,缓存只有可怜的16MB,根据下面的计算方法,可以基本肯定这是SMR盘了。但令人想不通的是,东芝这么抠搜的嘛,连大容量缓存都不舍得用啊。隔壁的希捷可都用上128MB了,32倍啊有木有,就算是这样,依然还是会卡。在这里需要提醒大家一下,目前市面上所有的7mm厚度的笔记本硬盘,超过1TB容量的几乎可以100%肯定是SMR盘。因为在2.5寸的体积和目前的技术限制下,传统PMR的单碟容量是不可能达到1TB的,而7mm内部空间非常紧凑,很难容纳两张碟片,所以厂商肯定会使用SMR来保证容量达到需求,至于性能,这几乎是无解的,只能期待SSD技术越来越先进,越来越便宜,逐渐普及并替代这些SMR盘吧。顺带一提,三星的QLC固态即将上市,大容量加上还算合理的价格(大约是1元1GB的水平),大家可以关注一下。磁盘占用100%扫描坏道中。每次扫描发现的坏道都不一样哦3000多小时就出坏道了,啧啧啧===20181104更新===还是这块盘,ST2000DM008,昨天把盘进行了全盘格式化,格式化成了exFAT格式,然后连到PS4上备份数据,PS4是最新6.02系统。总共700多GB的游戏本体+存档,4小时左右备份完成,并没有遇到占用100%的情况。为何会这么确定呢?因为后来恢复备份也是这个时间。推断大概是因为格式化之后,硬盘不再保留原来的FTL,并且整个备份过程应该是顺序写入的,不会出现覆盖磁轨的现象。但是要知道,这块盘从第一天买回来上机,在windows10上从旧盘往里拷贝文件时就出现了100%占用,那时候也是空盘啊,也不应该出现磁轨覆盖导致写入放大,该如何解释?恐怕只能怀疑windows10的缺陷或者和SMR不兼容造成的问题咯。是的,没错。SMR有天生的缺陷,写入时会覆盖临近磁轨的数据,要先把这些数据存到临近的缓冲区,然后写入新数据,再把旧数据写回去,所以可靠性肯定会下降,而且速度非常慢。为了缓解这种掉速,才用了256MB甚至更高的大缓存,而缓存本身是易失性的,一旦掉电就没了,很容易造成没来得及写入硬盘的数据丢失。科普视频:机械硬盘应该如何买?SMR叠瓦式硬盘是什么意思?【呼呼科普】_哔哩哔哩 (゜-゜)つロ 干杯~-bilibili科普文:详解叠瓦式磁记录SMR盘基础知识 - CSDN博客,SMR磁盘 - 随笔分类 - taoliu_alex - 博客园由于SMR的缺陷,SMR盘无法实现随机写入,因为改写一个文件,会影响后续的数据。所以就有了分块(band),一般一个band为256MB大小,设有缓冲区,改写时只会改这256MB的数据,不会影响别的数据,降低了存储密度,提升了可靠性。但这样性能仍然不可观。想一想,如果就更新1MB的数据,造成256MB的数据全部重写,这速度能快到哪去?为了解决随机写入的问题,就有了传统PMR缓冲区,这就类似于TLC的SSD为了解决写入速度问题而设立了SLC缓冲区,写数据时,先进缓存,然后写入PMR缓冲区,等写入结束了,硬盘空闲了,就把这些数据慢慢地搬到SMR区域。由于是机械硬盘,写入放大的影响不会像SSD那么致命。这样就必然会有一个逻辑层到物理层的映射机制,类似于SSD的FTL,它有可能影响寻道性能,并且它对整个硬盘的数据安全非常重要,一旦它出现问题,就有可能造成整个盘的数据不可识别,从而出现掉盘的现象,所以对数据恢复也是种考验。那么问题来了,这个PMR缓冲区设多少合适呢?目前没有这类资料。我手上有一块ST2000DM008,单碟2TB,正是用了SMR技术,同时还有ST2000DM006,双碟2TB传统PMR盘。在使用时,读取速度SMR要比PMR更快,但差距并不算很大。写入速度在数据量小的时候,也不会感觉到区别,甚至会觉得SMR速度更快一些。使用HDTune测速时,发现了非常诡异的现象。传统硬盘的速度一开始在200MB/s附近,越往后速度越慢,呈现一个下跌的趋势。但SMR盘会从一开始的200MB/s以上,在200GB左右时会出现一个断崖式下跌,速度可能会跌到1MB/s,然后在这里平稳一段之后,在400GB左右又会涨回200MB/s,往后的速度会出现数次往复,但始终都会在200MB/s附近,这种情况不知道是我的盘个例,还是因为有一定负载,还是普遍情况。最近发现一种说法,在新盘完全没拷入数据时,由于主控知道盘里哪里有数据哪里没有,那么在测速时,主控可能会不进行读盘而直接返回0,所以就算长时间测速,速度也不会有太大变化。那么主控怎么知道这个数据呢?必然不是全盘扫描得到。根据测试发现,用CrystalDiskInfo,在硬盘空闲时反复刷新磁盘SMART的时候,会听到磁头加载和卸载的声音,与此同时,C1也会同步加一,因此可以判断,这些区域使用的元数据(应该就是上面说的FTL映射机制),包括SMART数据,都是写在硬盘上的,在数据更新的时候,也会同步更新这些数据。那么,在对SMR盘测速时,可以猜测硬盘先读取这里的元数据,如果该位置没有数据(记为0),就直接返回0,速度就为元数据所在位置的速度;如果该位置有数据(记为1),就会移动磁头到对应位置读取对应的数据,速度就为对应位置的速度了。那么可想而知,新买的空盘,元数据全部为0的情况下,测速就是元数据所在区域的速度,但是当硬盘有一些数据,或者被填满的时候,速度因为要先读取元数据再寻道的原因,会比传统的直接寻道要慢。由于硬盘不同位置速度不一样,存的东西也各有不同,有的区域是空的,有的区域填满了数据,再加上硬盘有时候会有数据请求的负载,这也有可能是测速时急剧掉速的原因。因为此盘不在身边所以无法截图,贴一些网友的测试:西数SMR 2TB本盘,新盘测试补充一张惨得一皮的SMR盘测速。很明显,掉速的地方是有数据的ST2000DM008 SMR叠瓦式单碟2TB硬盘测速ST2000DM006 传统PMR双碟2TB硬盘测速ST1000DM010 传统PMR单碟1TB 有负载测试从上面几张图可以看出,在有负载的情况下,PMR单碟1T的盘很少会低于10MB/s的速度,但上面SMR单碟2T则出现了最低5.5MB/s的现象,结合我的盘测试结果,应该可以看出一些情况下确实会掉速,而且掉速非常严重。接下来说说长期困扰我的磁盘占用100%的问题。之前我笔记本自带1TB 7200转的硬盘,然后光驱位加了一块1TB 5400转的盘。升级到Windows10之后,使用一切正常,除了5400的盘略微慢一些之外,基本没有问题。但随着使用,数据量日益增长,笔记本没有额外的硬盘位了,于是想将5400的1TB盘升级成2TB,于是购买了希捷的7mm 2TB本盘ST2000LM015,测试一切正常,数据拷入就开始用,当天晚上用迅雷下载就出现占用100%的情况,不过这没持续多久就正常了。然后接下来噩梦就开始了。刚开始从1T迁过来的时候,速度一切正常。但当剩余空间就剩200GB时,这盘的写入速度就可以说是令人发指了,经常会掉到十几MB/s,而且这还和盘的剩余空间大小有关,剩余空间越小,写入就越慢,越有可能出现100%占用,一旦100%占用,基本读取速度掉到1MB/s,写入几百KB/s。本来以为是Windows10的锅,但不对啊,系统装在SSD上,从来没有过,应用软件装在1TB 7200盘上,也从来没有过100%占用啊,况且按照网上的教程,该改的都改了,该关的都关了,英特尔的RST驱动也是装了卸、卸了装,折腾N次没用。而且大部分情况下,关机再开都没用,开机直接100%占用没商量,就差重装系统了。注意哦,出现这种情况时,SMART的数据一切正常的哦,坏道扫描是全绿的哦。就这么折腾了几个月实在受不了了,换了块ST2000LX001,这是希捷酷玩混合盘哦,里面有8GB SSD加成哦。卡顿问题略有缓解,但还是经常出现100%占用,由于有SSD缓存,读取速度好了不少,写入速度,尤其是大文件写入速度还是令人发指的慢。作为日常装游戏、下载用的娱乐盘,速度总是慢人半拍。玩lol时,客户端(就是开始游戏选人的那个)载入转圈可以转5分钟,总是最后一个进入游戏,还时不时闪退,帧数经常徘徊在30~40左右。要知道,这电脑用的是960M的显卡,之前玩lol帧数可是能上100多的。检查了驱动、游戏设置均正常,帧数就是上不去,而且不仅仅是掉帧,有时候会直接卡住,完全动不了,持续十几秒,这时候切出去看,保准100%占用,响应时间上万毫秒。要知道,lol里这十几秒的卡顿会出现多少意外。同理还有GTA5,单机模式进游戏至少5分钟,OL模式更不用说,一旦队友掉线,那就慢慢在天上看云吧。至于下载,开迅雷100%是日常我会乱说。同时发现一个奇怪的现象,一旦出现100%占用,这时候打开hdtune进行坏道扫描,很快磁盘占用率就会下来,虽然还在90%以上,但读写速度均恢复正常。而一旦停止扫描,100%占用就会很快恢复。但这个方法并不是100%奏效,有时候就算开着坏道扫描,依然是100%占用,响应时间甚至会更长,上万甚至上十万毫秒。后来查资料得知,这俩盘都是希捷的SMR盘,要不2TB怎么可能做到7mm厚度?那么,为何平时用着都正常,为何会突然出现长时间100%占用呢?其实原因很简单。如果你玩SSD的话,TLC的盘,如果拷贝的文件超出SLC缓冲大小的话,速度就会直线下降,但SSD毕竟是SSD,就算掉速也比机械硬盘要快得多。而机械硬盘就没那么幸运了。上面说了,为了解决SMR不能随机写入的问题,会有一块PMR缓冲区,但这个缓冲区大小我不确定,大概是跟盘的设计容量和盘片大小有关。数据先写入PMR区域,等空闲时,再把这些数据搬到SMR区域存放,这个搬运的过程应该是非常耗费资源的,而在搬运过程中,如果有新的读写请求,就会出现掉速的现象。但是我想说的是,你并不知道硬盘固件会选择何时去搬数据,也不知道触发搬数据指令的条件是什么,那么就根据现有情况,大胆地猜测一下:硬盘闲置(没有或很少的读写操作)一段时间后,触发搬迁操作。这时候用户一般不会有感觉,除非硬盘声特别大。PMR区域占用达到一定比例时,触发搬迁操作。这时候如果没有数据读写,用户也感觉不到。PMR区域满了,有新数据存入,为了保护SMR区域的数据完整性,不能直接往SMR区域写入,那么就触发了搬迁操作,缓存写满后,限制写入速度,拷贝文件时,一开始会很快,但当缓存写满后,速度就会直线下降,任务管理器中查看硬盘出现100%占用。写入的数据超出PMR区域剩余空间时,缓存写满后,限制写入速度,同时触发搬迁操作,拷贝文件时,一开始会很快,但当缓存写满后,速度就会直线下降,任务管理器中查看硬盘出现100%占用。如果缓解了PMR空间危机,同时用户有大量读写请求时,暂停搬迁,直到读写结束后再恢复。如果触发了搬迁操作,那么无论是关机、重启后,搬迁操作均会继续,直到完成。搬迁进行中,如果强制关机,有可能造成数据损失,甚至损坏硬盘,造成不认盘(FTL挂掉)。那么就可得知,当磁盘占用100%时,操作系统并没有对硬盘做什么读写操作,任务管理器中硬盘的读写速度都很低,活动时间100%,平均响应时间从几百毫秒到数万毫秒不等。出现这种情况时,读取速度有时候能上数十MB/s,但写入速度基本上是小于10MB/s的,这时候就是硬盘在进行搬迁操作。那么既然这根操作系统无关,为何磁盘100%占用会集中在Windows10爆发呢?那是因为,SMR是近几年才开始流行的技术,而Windows7、Windows8时代还没有这种盘。到了Windows10时代,SMR技术被率先用在笔记本硬盘上,于是这种占用100%的问题开始凸显,尤其是比较低端的、没有SSD的笔记本上,单碟2TB的SMR盘也是今年才开始出货。至于网上流传的折腾驱动、安装快速存储技术等方法,其实可以理解为心理安慰。当你费心把原来的驱动卸了,装了新的驱动,经历了N次重启之后,可能这时候数据搬迁已经结束,占用率自然下来了。但是等过了几天,缓冲区又写满的时候,这种问题又再次重现。其实建议大家关闭系统的数据收集服务Connected User Experiences and Telemetry,它可以减轻硬盘的读写,稍稍避免频繁的100%占用。不过这个服务鸡贼得很,经常会偷偷地重启,要时不时去服务管理里看一眼。虽然SMR技术提升了硬盘容量,减轻了硬盘重量和功耗,但从实际使用体验可以发现,希捷的固件做得并不完善,它并没有及时地在空闲时进行数据搬迁,而是当PMR区域使用到一定比例时才会触发搬迁操作,但往往这时候都是在持续写入,于是就造成了掉速。但同时硬盘固件又有一个策略,那就是当硬盘内数据较少时,新增的数据会直接写入SMR区域的空闲区域,不会破坏临近磁道数据,同时也不经过PMR缓冲,这时候也不会感觉到掉速。但随着使用时间的增加,盘里的碎片越来越多,剩余空间越来越少,写入就越来越有机会写到有数据的磁道附近,而这种写入会破坏临近磁道数据,因此就不得不利用PMR缓冲,然后进行搬迁和垃圾清理,这就会造成掉速和卡顿。这就是为什么新买的盘速度很快,但用了一段时间后,数据越来越多时就容易卡顿的原因。当然,这跟主控的优化也脱不开关系。如果主控能及时地进行垃圾回收,优化数据搬迁的策略,并且开放上层接口,与操作系统结合,SMR盘的体验必然是会比现在更好的。这种盘买来当仓库盘,数据拷进去后就几乎不会再动,或者只会往里追加,没什么问题,甚至还可以感觉到大容量少盘片带来的好处,震动和发热大幅降低。但很多人买了这种盘来装系统装软件,殊不知系统的随机写入量是非常大的,各种软件的安装、注册表、设置、软件数据的保存、系统更新、日志等等,它对SMR盘的压力是很大的,所以说SMR这种盘绝对不适合装系统,不然就等着两小时的安装,两小时的更新,遇到大版本更新一下午都不一定能搞定。至于作为下载盘,写入量也是很大的,尤其是迅雷这种BT下载软件,会在开始下载时预申请空间,然后多线程填充数据,这个过程恰恰就是SMR盘最忌讳的,随机写入!剩余空间大了还好说,有PMR缓冲区挡着。如果盘都快满了,那就不要纠结为何下载会这么慢了,因为——你丫下得再多,我写不过来啊!注意哦,不要以为SSD有写入寿命,机械硬盘磁臂磁头也是有寿命的!磁臂的末端有一条排线,通过金属触点和外部电路板连接,这条柔性排线在磁头寻道运行时会发生形变,而上面的金属材料是有弯折次数限制的,这就是金属疲劳。一旦金属疲劳现象导致排线上的电路断连,就会导致磁头无法再使用。不过大部分硬盘都会考虑这种情况,寿命都会很长。现在的硬盘几乎都有斜坡加载技术,硬盘空闲、关机时磁头放在斜坡停泊区,需要读取数据时才会移到磁盘上。那么,问题来了,斜坡和盘片肯定不是一体的,每次磁头从盘片移到斜坡,或者移出,必然会出现形变,伴随着的就是偶尔会出现的嘀嗒声、咔嗒声和金属声。曾经西数盘出过卡顿门,原因就是固件过于激进,设定8秒没有读写操作就把磁头移回停泊区,每次进出就会增加SMART里的C1值,所以这批盘的C1都会暴涨。当有读写请求时,磁头移回盘片是需要时间的,甚至某些盘会降低转速节能,需要用到时再加速,这都会造成数据读写的延迟,造成卡顿。如果磁头在盘片上的时候突然断电,为了保护盘片,磁头会快速地回到停泊区。如果你拆过硬盘并做过实验的话,就会发现,这个归位的过程是非常快的,甚至归位时发出的声音也要比正常归位的声音要大很多,这是因为断电后盘片旋转速度下降,会导致磁头高度降低,而这时候再移回斜坡就相当于磁头被撞了一下,由于磁头这里的材料非常薄和脆弱,这个归位的动作很容易造成磁头变形,轻者能正常工作,但声音会变大,重则无法使用,甚至划伤盘片,造成不认盘。上述中的ST2000LX001这块盘,经常在使用时听到磁盘加速运转,然后咔嗒的声音,说明盘在偷懒,降低转速并磁头归位。但这块盘并没有感觉到明显的延迟和卡顿,为何?因为这盘自带8G的SSD加成,热点数据被放到SSD里,速度自然就快很多,而且如果在硬盘偷懒时,数据请求在缓存中命中的话,是不会有卡顿的。可想而知,如果没有这个SSD,这个加速运转和磁头移出,必然会造成卡顿。传统硬盘在数据写入时,数据写入结束后,通常不会再对这些数据进行别的操作。而SMR由于其特殊性,必然要对已写入的数据做二次转移的操作,并更新映射层FTL,而当文件数量很多的时候,可以很明显地听到磁头运转的声音。相同时间下,SMR的磁头明显要比传统硬盘运动量更大,做的操作更多,所以寿命肯定不会比传统硬盘更高。那么,重点来了:为什么硬盘商刻意隐瞒用SMR技术,并且将这种技术在低端市场推广呢?答案就是:省成本啊!!!!使用这种技术,盘体、磁头等不需要做多大改动,最重要的是固件的支持。然后呢?ST2000DM006和ST2000DM008这俩售价都是399,一个双碟4磁头,一个单碟双磁头,一个厚一个薄,价格不变物料成本就这么省下了,在寿命和性能以及售后的权衡之下,那自然是硬盘商赚得更多啊!至于性能,emmmmmmmmmmmmmmmmm有那么大的缓存和PMR缓冲区,还想怎样,低端硬盘,要啥自行车!事实证明,这种推广还是很有效果的,大缓存啊,好!单碟啊,功耗低很薄很轻,好!至于SMR?我不说你知道?最终上当受骗的还是买这种盘的消费级用户,至于企业?他们有更加靠谱的企业级盘,还会选这种?其实硬盘厂商应该反思一下,为什么隔壁固态硬盘都用上TLC甚至QLC了,人家也有SLC缓存,为啥不会出现这么大规模的掉速和长时间100%占用。当然,写入速度是一方面,另一方面还是看固件的调度。当硬盘空闲时,固件就应该及时地检测并在后台搬迁、清理这些数据,而不是当用户使用时,需要往里写入了,固件才发现缓冲区满了,再去搬家,这样效率是不是就会高很多。而根据我的观察,大部分硬盘空闲的时间里,磁头都是归位的,甚至盘都会减速运行,直到需要读写时才会恢复,很明显固件在数据管理方面还是非常怠惰的,并且节能机制非常的激进,几秒到几十秒没有读写操作,就会把磁头归位。虽然这对硬盘来说是一种保护,但对SMR这种盘来说,却是造成性能下降、100%占用的罪魁祸首。不过顺便一提的是,NAS盘和监控盘,由于其对随机写入的性能要求很高,尤其是监控盘,几乎是24小时都在连续进行读写工作,这种压力下根本没时间对硬盘数据进行回收迁移等操作,所以这种盘一般不会使用SMR技术。另一方面,由于SMR技术必须要求至少256MB的缓存,所以在选购硬盘时,选择低于256MB缓存的硬盘,也是个很好的选择,但是大缓存的性能优势也是不能忽视的,尤其是对大容量硬盘而言。不过有一个更高端的方法,那就是看硬盘商的硬盘参数,重点看盘片数和总容量,计算单碟容量。目前传统的PMR盘,3.5寸单碟容量极限在1.25~1.5TB左右,2.5寸单碟容量极限在500~750GB左右,一般情况下单碟容量超过这个值的肯定是SMR盘无疑了。编辑于 2019-11-08 13:26赞同 866194 条评论分享收藏喜欢收起匿名用户题主自问自答一下……太长不看版:SMR是坑爹玩意儿,从道理上讲,能绕开最好绕开,但它还是未来的趋势,不太好绕开……哎。至于官方质保,并不包括数据恢复。不管怎样,自己多备份几份肯定更好。还有各种缓存加速,比如傲腾、SSHD等,个人不看好,因为缓存的加速原理是把经常访问的热数据放到读写相对极快的闪存/DRAM中,从而避免执行慢吞吞的读盘/写盘操作;而缓存容量是极其有限的,如果热数据撑爆了缓存/大量写入数据,SMR的掉速问题就会暴露无遗;而且缓存也会让系统变得更复杂,所以暗藏bug的概率也会变大。先扣一下字眼:为什么大家都在说SMR vs PMR呢?好像应该是LMR vs PMR、CMR vs SMR这样才对吧。可能是因为PMR正好是SMR之前的上一代技术改进?LMR:Longitudinal Magnetic Recording,水平磁记录;PMR:Perpendicular Magnetic Recording,垂直磁记录;对比图,来自维基百科:CMR:Conventional Magnetic Recording,传统磁记录;SMR:Shingled Magnetic Recording,叠瓦式磁记录。对比图,来自希捷官网:如何判断一块盘是不是SMR?1.从这个帖子里似乎可以看到:新出厂的SMR空盘会跑出很平直的读取速度曲线和很低的存取延迟。西部2T新2.5寸盘有点厉害啊 - 电脑讨论 - Chiphell - 分享与交流用户体验如何解释呢?其实很容易脑补出来:硬盘主控知晓哪些区域是空的,所以不必执行读盘动作,直接给主机返回一串0即可。所以,我们就看到了平直的“高速读取”曲线和低得吓人的读取操作延迟——因为压根就没执行读取嘛。原帖里有人说是缓存,答主觉得和缓存基本无关,因为很显然,0.5GB都不到的缓存是没法对2TB的顺序数据读取操作产生什么影响的。对于用了一段时间、里面已经写了不少数据的SMR盘来说,测出来的读取速度曲线就类似非SMR的硬盘了,已经有答主贴出了截图。2.SMR盘往往搭配128MB以上的大缓存。3.SMR盘往往盘体更薄。4.SMR盘单碟容量更高——不过这个貌似只能大概推断一下,需要上网搜吧。貌似没了……比如价格方面,两者看上去好像差不多……其次,SMR盘有什么问题?1.“脏盘掉速”——写入比较多的数据后,读写速度会突降SMR盘也可以拥有和非SMR盘相仿的顺序写入性能,但是因为写入时会覆盖邻近磁轨的原因,不仅随机读写性能会跪,顺序写入性能也会跪……2.性能时好时坏、不可预测,而且大体上是走下坡路的显然,有的时候邻近的磁轨是空的,所以可以直接写、不用担心覆盖破坏数据;有时候就不是这样了。而且磁盘空间里保存的数据越多,覆盖邻近磁轨的机会就越多,需要反复折腾回写的时候也会越多。题主原以为只有写入会掉速,不过看楼上的情况,貌似不仅写入会受影响,读取可能也会受影响——这可能是硬盘主控在后台执行类似SSD垃圾回收的“数据搬运”动作导致的?3.缺乏操作系统层面的优化支持众所周知,老的操作系统如WinXP缺乏TRIM支持,导致SSD出现使用一段时间后严重掉速的问题。没想到机械硬盘现在也面临类似的蛋疼情况:想写入时不能直接写,需要反复折腾回写,“写入放大”……按照这个逻辑,SMR硬盘即使是用作保存影音文件冷备份等顺序读写、写少读多的用途,仍然可能碰到严重掉速等问题,因为主控并不知道哪些数据已经被丢弃、可以被直接覆盖掉,做了很多无用功;操作系统也不顾在哪里写入对性能的影响最小,只顾着按照非SMR盘的老习惯往里写……4.可靠性存疑多次回写是否会迫使磁头频繁移动、导致损耗加速?回写过程中万一断电了,主控可以搞得定么?会不会导致数据损坏?会不会发生类似“Intel 8M门”那种主控bug事件?消费者应该怎么办?1.明确自己的需求,对于装系统、软件、游戏、P2P下载等需要随机读写性能的用途,绕开SMR盘。最适合SMR的是顺序写入、写少读多的存档用途,比如各种影音数据、整盘备份等数据。对于NAS、RAID阵列等用途答主不太确定,不过答主觉得最好还是避开SMR。2.等待操作系统(文件系统)对SMR盘的识别和优化技术普及,也就是Host-Aware SMR和Host-Managed SMR。目前的SMR多是为了照顾兼容而舍弃性能的Drive-Managed SMR,操作系统不知道这块盘是不是SMR的,所以还是按照非SMR的方式来使用、管理它,信息不能沟通,自然会带来工作效率下降的问题。3.避免进行碎片整理。碎片整理需要进行大量的随机读写操作,对SMR本来就很孱弱的随机读写性能来说实在是雪上加霜。4.做好备份工作。鸡蛋放在一个篮子里当然不太保险。而且……官方质保也不保护你的数据。emmm……其实答主自己就没做好这块……不过备份工作应该做好,这个是大家都认同的共识对吧( ̄y▽, ̄)╭ 编辑于 2018-09-12 20:11赞同 15224 条评论分享收藏喜欢
叠瓦磁记录 - 维基百科,自由的百科全书
叠瓦磁记录 - 维基百科,自由的百科全书
跳转到内容
主菜单
主菜单
移至侧栏
隐藏
导航
首页分类索引特色内容新闻动态最近更改随机条目资助维基百科
帮助
帮助维基社群方针与指引互助客栈知识问答字词转换IRC即时聊天联络我们关于维基百科
语言
语言链接位于页面顶部,标题的另一侧。
搜索
搜索
创建账号
登录
个人工具
创建账号 登录
未登录编辑者的页面 了解详情
贡献讨论
目录
移至侧栏
隐藏
序言
1历史
2数据管理
开关数据管理子章节
2.1由设备管理
2.2由主机管理
2.3主机可感知
3协议
4软件及应用
开关软件及应用子章节
4.1动态混合SMR
5参考资料
6参见
7外部链接
开关目录
叠瓦磁记录
7种语言
DeutschEnglishFrançaisItaliano日本語Norsk bokmålPolski
编辑链接
条目讨论
不转换
不转换简体繁體大陆简体香港繁體澳門繁體大马简体新加坡简体臺灣正體
阅读编辑查看历史
工具
工具
移至侧栏
隐藏
操作
阅读编辑查看历史
常规
链入页面相关更改上传文件特殊页面固定链接页面信息引用本页获取短URL下载二维码维基数据项目
打印/导出
下载为PDF打印页面
维基百科,自由的百科全书
此條目翻譯品質不佳。 (2020年6月4日)翻譯者可能不熟悉中文或原文語言,也可能使用了機器翻譯。請協助翻譯本條目或重新編寫,并注意避免翻译腔的问题。明顯拙劣的翻譯請改掛{{d|G13}}提交刪除。
随机写入SMR较为困难:写入一个磁道会覆盖下一个磁道上的数据。必須要有管理系統來避免重複寫入。
叠瓦磁记录(英語:Shingled magnetic recording,SMR,直译为分层磁记录),是一种用于硬盘驱动器的磁存储数据记录技术,可提高存储密度和每个驱动器的整体存储容量。[1] 常规的硬盘驱动器通过写入彼此平行而不重叠的磁道来记录数据(垂直磁记录,PMR)。而叠瓦磁记录技术的硬盘写入的新磁道则与先前写入的磁道部分重叠,从而使先前的磁道更窄,因此能拥有更高的磁道密度。由此可以看出,使用叠瓦磁技术的磁道相互重叠,与用作屋顶的瓦片堆叠方式类似。我们之所以能这样做,是因为磁盘写入磁头由于物理上的原因比读取磁头宽上许多,因而由正常方式写入的磁道宽度远比读取磁头所需的磁道宽度来得宽。[2][3][4]
由于磁道存在重叠,叠瓦磁盘的写入过程较为复杂。如果我们随机写入一个磁道,由于写入磁头的宽度比磁道宽,因此写入会影响到临近磁道;如果这个临近磁道有数据,这些数据就也需要重写以免数据被破坏,依此类推。因此,SMR 磁盘一般分成很多块只能追加数据(顺序写入)的区域(Zone),这和固态硬盘的闪存页管理类似。使用“由设备管理”(device-managed)方式的 SMR 磁盘通过内部固件处理了 SMR 磁盘复杂的写入问题,从而对使用者封装了 SMR 磁盘的复杂性,令使用者可以像使用 PMR 硬盘一样随机写入 SMR 硬盘。其他 SMR 磁盘则使用“由主机管理”(host-managed)方式,需要操作系统识别 SMR 磁盘并拥有能对 SMR 磁盘进行正确顺序写入的驱动程序才能被正常使用。[4][5]
历史[编辑]
希捷科技从2013年开始出售“由设备管理”的 SMR 磁盘,并声称使用 SMR 技术的磁盘总体容量比普通磁盘的高出25%。[1]2014年,HGST推出了一个容量达10 TB的 SMR 氦气硬盘,[6]不过到2015年12月又推出了10TB的 PMR 氦气硬盘。[7]2018年11月,HGST 推出了14 TB和15 TB的 SMR 硬盘。[8]
鑒於SMR的複雜性,威騰電子、希捷、东芝都曾在或仍在未标明磁盘是否使用 SMR 技术的情况下出售硬盘。这导致了很大的争议:因为 SMR 硬盘在一些情况下显著慢于 PMR 硬盘。[9]无论是面向数据存储(服务器、NAS、冷存储)的硬盘以及面向普通消费者的硬盘有这个问题。由于一些 NAS 设备在使用 SMR 硬盘后出现数据损坏,导致消费者投诉,主要硬盘品牌最终公布了使用 SMR 技术的硬盘产品型号并保证特定系列型号不使用 SMR 技术。[10][11]
数据管理[编辑]
SMR 盘上的数据有3种管理模式:[12][13]
由设备管理(device managed)
由主机管理(host managed)
主机可感知(host aware)
由设备管理[编辑]
由设备管理的 SMR 硬盘在接口上对主机来说和一般磁盘没有区别。主机不需要使用任何特殊方式来写入磁盘,并且也不需要知道磁盘是否使用了叠瓦存储技术。其连续写入性能较随机写入高。一切关于数据位置的处理由磁盘本身管理。[4]
这类 SMR 硬盘的固件控制机理和固态硬盘类似:主机所使用的 LBA 地址和磁盘实际的物理结构没有直接关系,而是在固件中存在一个“转换层”对两者进行转换。由于在只能追加(顺序写入)的区域里进行随机写入非常慢,所以这类硬盘都会先把数据写到一个 PMR 缓存区,等到空闲的时候再将数据整理进 SMR 区域中。由于硬盘在处理 RAID 重新同步时的写入次数很多,这使得在 RAID 中使用 SMR 硬盘容易使缓存超出,进而导致 SMR 盘不时需要暂停数分钟进行整理。还有些问题固件(如 WD40EFAX)会拒绝读取没有写过的 LBA 地址,并向调用者报告一个错误。SMR 硬盘的这两种处理方式都会导致 RAID 控制器认为磁盘损坏。[14]
SMR 硬盘的分块结构和固件的垃圾回收机制也意味着磁盘存在写入放大的问题,[15]不过这对于硬盘来说写入导致的主要是速度而不是寿命问题。有些 SMR 盘支持Trim命令,以便避免整理不需要的空间。[16]
由主机管理[编辑]
由主机管理的 SMR 硬盘需要主机严格按照特别的协议流程来写入数据。主机必须顺序写入,以避免破坏已有的数据。磁盘会拒绝执行违反这一协议的命令。[4]
主机可感知[编辑]
主机可感知是由设备管理和由主机管理两者的组合。磁盘可以处理任意顺序的写入命令,但是主机可以知道磁盘使用了叠瓦存储,也能知道具体的叠瓦空间使用情况。一方面新主机可以尽量顺序写入提高性能,另一方面对旧系统也保持了兼容。[4]
协议[编辑]
SMR 设备在协议中称为“分区域块设备”(zoned block device),每个区域的大小一般为 256MiB。[17]在由主机管理和主机可了解的 SMR 盘上,SCSI 的 ZBC 和 SATA 的 ZAC 这两套命令会暴露给主机。这些命令可以让主机了解每个区域的储存科技(SMR/PMR),并对这些区域直接进行访问。[18] ZBC/ZAC的命令有:
报告区域 REPORT ZONES,用来获得磁盘区域布局和区域情况(包括表示每个顺序区域写了多少的写入指针)
SMR这类的区域在由主机管理的磁盘上会标为“必须顺序”(sequential required),在主机可了解的磁盘上则是“偏好顺序”(sequential preferred)
重设写入指针 RESET WRITE POINTER,用来归零指针,清空顺序区域
打开区域 OPEN ZONE,用来给区域“加锁”,以便独占访问
关闭区域 CLOSE ZONE,用来解锁区域
收尾区域 FINISH ZONE,用来填充区域,并标记为可读
每个区域都有自己的LBA范围。只要遵守了“必须顺序”区域的规定,所有一般的 LBA 命令都可以使用。
SMR 磁盘可以通过以下的方式报告自己的分区域属性:[19][20][4]:14
由设备管理和主机可了解的磁盘会有一般块设备(SCSI 00h)的标记,方便一般计算机识别。
一个叫做ZONED的字段会显示设备是由主机管理,主机可了解,还是都不是。这个字段处于 SCSI 的块设备特性(Block Device Characteristics)VPD 页面和 ATA 的设备能力日志页。
由主机管理的块设备有一个新的设备种类标记(SCSI 14h)。只有支持这些协议的电脑才可以识别。
这套标准的第二版称为 ZAC-2/ZBC-2,截至2020年4月 (2020-04)[update]仍在开发中。这类设备引入了一种“领域和范围式分区域块设备”(domains and realms zoned block device),支持不连续的 LBA 地址。[21]西部数据的代表提议,要在新标准里移除对 ZONED 字段的描述。[22]
这套分区系统可以对闪存也有意义,有助于降低内部控制器的工作需求,优化写入放大并降低预留空间。NVMe组织也已提供一个类似的接口,称作 ZNS。[23]
软件及应用[编辑]
叠瓦磁记录的属性介于只能顺序读写的磁带和可以随机的读写的普通硬盘之间:其数据密度较高,可以随机读取,但只能顺序写入。这类硬盘适合写入一般不需要回头修改,但需要经常随机读取的数据。Dropbox 的“魔术口袋”(Magic Pocket)就是这样一种系统,它把用户的数据存储在只能向后添加的文件里。[24]也有些由设备管理的 SMR 硬盘被当作“存档用磁盘”售卖。[25]
Linux有一些文件系统已经支持 SMR 设备或者可以调整得性能更好:[26]
F2FS 本来为闪存设计,现在也有一个分区域块设备(Zoned Block Device, ZBD)的模式。可以在由主机管理的磁盘上使用,不过需要一块普通区域来随机读写元数据。
Btrfs 的 ZBD 支持在 5.12 内核添加,不过这个寫入時複製的文件系统本来就基本上进行顺序写入了。
有调整 Ext4 使其更多进行顺序写入的实验。
对于其他文件系统,需要使用 Linux 设备映射器(英语:device mapper)的 dm-zoned 目标把由主机管理的硬盘映射成一个普通的随机写入硬盘。4.10以上版本的内核不需要 dm 就可以自行映射。[27]
FreeBSD 也有对分区域块设备的协议级支持。[17]截至2020年4月 (2020-04)[update],Windows 和 macOS 都没有支持这些协议。
动态混合SMR[编辑]
传统的 SMR 盘是在生产时就规定了每一个分块的用途,但新兴的动态混合 SMR 盘可以由顾客随时重新指定分块是当 SMR 用还是当 PMR 用。[28][29]
参考资料[编辑]
^ 1.0 1.1 Anand Lal Shimpi. Seagate to Ship 5TB HDD in 2014 using Shingled Magnetic Recording. AnandTech. September 9, 2013 [February 9, 2015]. (原始内容存档于2020-06-04).
^ Roger Wood. Shingled Magnetic Recording and Two-Dimensional Magnetic Recording (PDF). ewh.ieee.org. October 19, 2010 [December 14, 2014]. (原始内容 (PDF)存档于2014-10-04).
^ What is Shingled Magnetic Recording (SMR)?. storagereview.com. January 30, 2015 [February 9, 2015]. (原始内容存档于2015-02-09).
^ 4.0 4.1 4.2 4.3 4.4 4.5 Mary Dunn; Timothy Feldman. Shingled Magnetic Recording: Models, Standardization, and Applications (PDF). Storage Networking Industry Association. September 22, 2014 [February 9, 2015]. (原始内容 (PDF)存档于2020-06-16).
^ Jake Edge. Support for shingled magnetic recording devices. LWN.net. March 26, 2014 [December 14, 2014]. (原始内容存档于2015-02-02).
^ Geoff Gasior. Shingled platters breathe helium inside HGST's 10TB hard drive. The Tech Report. September 9, 2014 [February 9, 2015]. (原始内容存档于2019-03-16).
^ Sebastian Anthony. HGST releases helium-filled 10TB hard drive; Seagate twiddles shingled fingers. Ars Technica. 3 December 2015 [3 December 2015]. (原始内容存档于2016-07-16).
^ 15TB and 14TB SMR Hard Drives Ultrastar DC HC620. www.hgst.com. [30 October 2018]. (原始内容存档于2018-10-31) (英语).
^ Alcorn, Paul. Sneaky Marketing Redux: Toshiba, Seagate Shipping Slower SMR Drives Without Disclosure, Too. Tom’s Hardware . [17 April 2020].
^ 新浪科技综合. 东芝官方公布 SMR 硬盘完整名单:桌面/笔记本五大系列. tech.sina.com.cn. 2020-04-29 [2020-05-23].
^ 避坑:希捷和东芝硬盘中的SMR型号_PCEVA,PC绝对领域,探寻真正的电脑知识. www.pceva.com.cn. [2020-05-23].
^ Zoned Block Commands (ZBC) (PDF). t10.org. ANSI T10 Committee. [22 January 2018]. (原始内容存档 (PDF)于2020-07-03).
^ Campello, Jorge. SMR: The Next Generation of Storage Technology (PDF). 24 September 2015 [22 January 2018]. (原始内容存档 (PDF)于2020-06-16).
^ Mellor, Chris. Shingled hard drives have non-shingled zones for caching writes. Blocks and Files. 15 April 2020 [2020-04-23]. (原始内容存档于2020-04-23).
^ Brewer, Eric; Ying, Lawrence; Greenfield, Lawrence; Cypher, Robert; T'so, Theodore. Disks for Data Centers. Proceedings of USENIX FAST 2016. 2016 [2020-04-23]. (原始内容存档于2020-03-10) (英语).
^ TRIM Command Support for WD External Drives. WD support. [2020-04-23]. (原始内容存档于2020-04-17).
^ 17.0 17.1 zonectl(8) – FreeBSD系统管理(System Manager's)手册页
^ SMR (Shingled Magnetic Recording) 101. Tom's IT Pro. [2018-03-03]. (原始内容存档于2017-06-11) (英语).
^ Information technology - ATA Command Set - 4 (ACS-4), Draft revision 18 (PDF). [2020-04-25]. (原始内容存档 (PDF)于2018-11-23).
^ Seagate. SCSI Commands Reference Manual, Rev. J (PDF): 472. [2020-04-25]. (原始内容存档 (PDF)于2020-03-12).
^ T10, 2020.
^ Weber, Ralph O. SBC-5, ZBC-2: Obsolete the ZONED field (PDF). www.t10.org. April 23, 2020 [2020-04-25]. (原始内容存档 (PDF)于2020-04-24).
^ NVMe Command Set Specifications. [2022-11-15]. (原始内容存档于2023-01-28).
^ Magic Pocket Hardware Engineering Teams. Extending Magic Pocket Innovation with the first petabyte scale SMR drive deployment. dropbox.tech. [2020-04-23]. (原始内容存档于2020-04-18).
^ Archive HDD (PDF). Seagate. [2018-03-03]. (原始内容存档 (PDF)于2017-10-11) (英语).
^ File Systems. ZonedStorage.io. [2020-04-23]. (原始内容存档于2020-01-29).
^ Device Mapper. ZonedStorage.io. [2020-04-23]. (原始内容存档于2020-05-22).
^ Collins, Brendan. Dynamic Hybrid SMR. Western Digital. 13 November 2017 [25 August 2018]. (原始内容存档于2018-08-26).
^ Dynamic Hybrid-SMR: an OCP proposal to improve data center disk drives. blog.google. Google. 13 November 2017 [22 January 2018]. (原始内容存档于2018-05-29).
参见 [编辑]
熱輔助磁性錄寫 (HAMR)
陣列媒體(英语:Patterned media)
二维磁记录(英语:Two-dimensional magnetic recording)
日志结构文件系统(英语:Log-structured file system),一类为仅添加存储设备优化的文件系统
外部链接[编辑]
LSFMM: A storage technology update(页面存档备份,存于互联网档案馆), LWN.net, April 23, 2013, by Jonathan Corbet
SMR Impact on Linux Storage Subsystem(页面存档备份,存于互联网档案馆), HGST, 2014, by Jorge Campello and Adam Manzanares
Layout optimisation for using XFS on host-managed SMR drives(页面存档备份,存于互联网档案馆), March 2015
SMR in Linux Systems(页面存档备份,存于互联网档案馆), Seagate, March 18, 2015, by Adrian Palmer
Host-Aware SMR(页面存档备份,存于互联网档案馆), Seagate, November 10, 2014, by Timothy Feldman
部分硬盘制造商、NAS制造商和科技社群公布的使用叠瓦磁记录技术的硬盘列表:
On WD Red NAS Drives(页面存档备份,存于互联网档案馆), Western Digital, April 22, 2020
Use of Shingled Magnetic Recording (SMR) technology in Toshiba Consumer Hard Drives(页面存档备份,存于互联网档案馆), Toshiba, April 28, 2020
List of known SMR drives(页面存档备份,存于互联网档案馆), iXsystems Forums, April 16, 2020
Synology Products Compatibility List (pre-filtered to SMR HDD), Synology
爭議事件
WD 在紅標 NAS 硬碟上使用 SMR 技術,國外消費者準備集體提告 | T客邦(页面存档备份,存于互联网档案馆)
取自“https://zh.wikipedia.org/w/index.php?title=叠瓦磁记录&oldid=80382615”
分类:電腦儲存技術硬盘驱动器隐藏分类:CS1英语来源 (en)自2020年6月粗劣翻译含有英語的條目
本页面最后修订于2024年1月5日 (星期五) 23:53。
本站的全部文字在知识共享 署名-相同方式共享 4.0协议之条款下提供,附加条款亦可能应用。(请参阅使用条款)
Wikipedia®和维基百科标志是维基媒体基金会的注册商标;维基™是维基媒体基金会的商标。
维基媒体基金会是按美国国內稅收法501(c)(3)登记的非营利慈善机构。
隐私政策
关于维基百科
免责声明
行为准则
开发者
统计
Cookie声明
手机版视图
开关有限宽度模式
机械硬盘避坑大法:一文搞懂PMR和SMR有什么区别
机械硬盘避坑大法:一文搞懂PMR和SMR有什么区别
行业咨询
品牌营销
集微资讯
知识产权
集微职场
集微投融资
搜索
爱集微APP下载
扫码下载APP
资讯
集微报告
舆情
JiweiGPT
2024半导体投资年会
集微视频
我的收藏
我的评论
我的消息
我的会议
我的报告
个人设置
舆情设置
安全设置
退出登录
登录
机械硬盘避坑大法:一文搞懂PMR和SMR有什么区别
作者:
爱集微
2019-07-31
相关舆情
AI解读文章
来源:IT之家
#PMR#
#SMR#
评论
收藏
点赞
21.2w
存储市场上一直存在固态硬盘(SSD)和机械硬盘(HDD)的竞争。论综合性能,SSD远高于HDD,是大家选购存储设备时的理想选择。早期消费级SSD存储容量一般不高,并且价格昂贵,让很多消费者望而却步。不过这两年,消费级SSD的存储容量不断提高,目前市场上也有4TB的产品可选;同时SSD的价格也在不断下跌,眼下采用原厂TLC颗粒的500GB固态硬盘售价也降到了300多元。SSD的这些进步令HDD的处境越来越尴尬。不过,尽管SSD取代HDD的声音越来越大,但在存储容量以及价格上的客观优势,HDD还是有的,并且它越来越偏向于被网友当做长期备份和存储数据资料的数据仓库来使用。在今天数据信息爆炸的年代,相信很多人都有大量可能不经常使用但又舍不得删除的资料需要长期保存,这些资料需要单独的硬盘来存储,机械硬盘,目前来讲是存放这些资料的最合适的选择。在这种场景下,硬盘容量的重要性很高,而在给HDD扩容的道路上,厂商们做过很多尝试,其中有PMR和SMR的区别。或许很多小伙伴对此并不了解,今天IT之家就为大家介绍一下机械硬盘PMR和SMR两种技术的区别。1、机械硬盘运行的原理目前很少有厂家会在机械硬盘的产品包装中注明该产品采用的是PMR还是SMR技术,对于HDD来说,这是一项比较深的技术参数,但是如果消费者购买不当的话,在一些使用场景下还是会比较坑的。而如果想更清楚地了解PMR以及SMR的区别,还是要从机械硬盘基本的结构原理说起。如图,是一个机械硬盘的内部结构示意图,它的主要部件包括主轴、磁盘、磁头,其他部件包括空气过滤片、音圈马达、永磁铁等。其中,主轴下方包含马达电机以及轴承;磁盘又被称作盘片,多采用铝合金材料,被固定在主轴电机的转轴上,工作的时候磁盘会随着主轴进行高速旋转,并且通常硬盘内的盘片数量都不止一片,当然,也不会很多。磁盘是用来存储数据的,具体如何存储数据,是我们后文要说的重点。磁头和磁头臂是是一个整体,磁头主要负责读写数据,在硬盘驱动器的控制下,磁头工作时会在盘面上快速移动,准确定位到指令要求定位的磁盘磁道上。这三者是硬盘能够读取、存储数据的关键,而其中关键中的关键,就是磁盘。磁盘的外观和我们见过的光盘类似,是数据的载体,因此我们有必要了解其内部数据的组织和管理结构。我们以单一的磁盘来看,它被划分为由一圈一圈同心圆组成的磁道,当然,这些磁道窄而密集,通常一个盘面就有上千条磁道。这些磁道肉眼显然是看不到的,但我们可以脑补它确实可以在盘面上看到:我们用简单的图例来表示,在下面这张图中,两个同心圆中空白的部分就是磁道,你可以理解为学校操场上的跑道。磁盘最外围的磁道我们称为0磁道,硬盘数据的存放就是从最外围的0磁道开始的;由此向内数,下一个磁道就是1磁道,然后是2磁道……同时,这些由同心圆组成的磁道并不是连续的,它们被横向地划分成了一道一道的圆弧,每一段磁道形成的圆弧,就叫做扇区,而在同一个圆心角范围内的扇区组成了一个扇面。具体在上面的图片中可以清楚看到。扇区是操作系统在硬盘上存储信息的具体形式,一个扇区包括512个字节的数据和其他的标记信息,例如标记扇区三维地址的信息方便寻址,还有“不良扇区”的标志等等。这里还有一个概念,就是柱面。我们刚才说过,一个硬盘中的磁盘通常不止一个,并且这些磁盘规格以及磁道分布都是一样的,所以,不同盘面上的同一磁道,可以构成一个圆柱,这个柱体就叫做柱面。IT之家这里就不针对柱面展开细说了,大家之需要知道,数据的读取和写入都是按柱面的顺序进行的,而不是按照盘面顺序就行了。接下来就是磁头了,它是硬盘读写信息的关键部件,主要作用,就是将存储在硬盘盘片上的磁信息转化为电信号向外传输。磁盘,也就是盘片,为什么能够存储信息?这其实和磁带的原理比较相似,在磁盘的表面,涂有一层薄薄的磁性材料,磁盘本身是铝合金材质,也有企业尝试过玻璃材质,磁性材料在磁盘表面可以涂敷得非常平整。而磁头,通俗来讲是用线圈缠绕在磁芯上制成的,写入数据的时候,磁头上的线圈通电,在周围产生磁场。高中物理中学过,改变电流的方向,磁场的方向也会改变,而磁场会磁化磁盘表面的磁性物质,使它们按照磁场的方向排列。切换不同的磁场方向,不同的磁性微粒也会有不同的方向,就可以用来表示“0”和“1”,我们知道,计算机中的数据都是以二进制的形式存在的,恰好,可以用这个方法来写入二进制的原始数据。同理,读取数据的时候,磁头线圈切割磁场线产生感应电流,磁性材料的磁场方向不同,所以产生的感应电流方向也不同,磁头就可以通过感应旋转的盘片上磁场的变化来读取数据。基本的原理很简单,但实际操作起来当然要求是需要很高的。首先,磁头需要采用特别的材料制作,因为它需要对磁感应非常敏感,并且要求极高的精密度,因此磁头的制造工艺和材料都不能随意;其次,硬盘工作时,磁头是不与高速旋转的磁盘表面接触的,而是以非常微小的距离飞行在磁盘表面,这样一来可以不让磁头擦伤盘面的磁性涂层,同时也不让磁性涂层损伤磁头;还有就是,在这种高速、精密的运转状态下,必须保证高度无尘,一旦进入灰尘,就有可能碰伤磁头或者划伤磁盘表面的磁性涂层,导致硬盘数据丢失甚至损坏。所以通常硬盘的内部都是密封的,在前面硬盘结构的图示中,我们也看到其外围还有一层空气过滤片。2、PMR和SMR技术的区别对于机械硬盘而言,容量的需求很高,怎样提高硬盘的容量呢?这就要回到机械硬盘存储数据的原理了。IT之家在上一部分已经讲过,数据是存放在硬盘内部一张一张磁盘盘片上的,具体是存储在盘片磁道上的扇区中。所以提升硬盘的整体容量有三个方法:第一是增加磁盘的数量,第二是增加磁盘的面积,第三是增加每个磁盘上存储数据的密度。前面两种方法势必会令硬盘整体体积增加,现代计算机硬盘的标准规格是3.5英寸,还有2.5英寸笔记本硬盘也比较普遍,另外还有用于超薄笔记本电脑的1.8英寸微型硬盘、1.3英寸微型硬盘等等,硬盘的尺寸规格是标准化的,随意增大或减小都可能带来不利影响。再进一步,硬盘内的盘片也不是越大越好,越大的磁盘,高速旋转时惯性越大,稳定性越低,所以转速上不去。所以,增加硬盘容量,最好的方法似乎是提升单个磁盘数据存储的密度。为了实现这个目的,硬盘厂商工程师们想了很多办法。我们已经知道,磁头通过感应盘片上磁场的变化来读取数据;通过改变盘片上的磁场来写入数据,以磁场方向的不同来记录0和1。在早期,磁盘上每个存储位的磁性粒子是平铺在盘面上的,磁感应的方向也是水平的。这种感应记录方式被称为LMR(Longitudinal magnetic recording),也就是水平磁性记录,这种方式有一个缺点,就是比较占面积,另外当磁粒过小,相互靠得太近,磁性就很容易受到热能的干扰,令方向发生混乱。所以,LMR的时代,单个磁盘能够存储的数据有限,整个硬盘的容量也就存在瓶颈。为了解决这个问题,后来人们想了一个办法,原来磁感应的方向不是水平的吗?如果让磁性粒子和磁感应的方向相对盘片垂直,这样不就能腾出很多空间了?于是人们发明了这种垂直磁性记录的方法,叫做PMR(Perpendicular Magnetic Recording),在此基础上,科学家还利用了热辅助磁记录技术,来提高在高密度下的信息写入能力。这种技术采用了一种热稳定记录介质,通过在局部进行激光加热,来短暂减小磁阻力,从而有效提高磁头在微场强条件下的高密度信息写入能力。▲gif来源:Youtube视频博主TED-Ed在PMR技术的帮助下,硬盘的存储容量得到了很大的提升,3.5英寸的硬盘,单碟磁盘的容量高可达1TB左右,这本质上是磁盘内信息存储的密度大大提升。不过随着互联网信息技术的飞速发展,信息数量爆炸式增长,人们要存储的东西也越来越多,渐渐的,PMR技术的硬盘,容量也不大够用了。怎么办呢?还有没有办法进一步提高磁盘信息记录的密度?有。不过这次科学家们想出来的办法有些奇特,并且也不像PMR那样完美,就是Shingled Magneting Recording(SMR)技术,又叫叠瓦式磁记录技术。这项技术是怎么做的呢?前面我们说到,磁盘是被划分为一圈一圈微小的磁道来记录数据的,这些磁道之间并不是连续的,而是磁道与磁道之间存在一个保护距离,从而不让不同磁道的数据产生干扰。硬盘工作的过程也就是磁头在磁道上读取和写入数据的过程。不过,现实中有一个情况,就是硬盘信息的读取和写入是两种不同的操作,所以读取磁头和写入磁头也是不一样的。现代硬盘主要采用的是分离式磁头结构,写入磁头仍是传统的磁感应磁头,比较宽,读取磁头则是新型的MR磁头(磁阻磁头),比较窄,磁道在划分的时候,当然要满足最宽的标准。但是写入磁头在工作的时候,实际上对于每个磁道,其写入信息的宽度是和读取的宽度一样的,这样,磁道的空间就存在浪费的情况。怎么解决这个问题呢?科学家们想到了一个“极限操作”,他们将磁道“被浪费”的一小部分重叠起来,就像咱们屋顶上叠加的瓦片一样。写入的时候沿着每条磁道上方进行写入,中间留下一小段保护距离(保护距离其实也缩小了),再写下一条磁道。如此一来,磁盘上磁道的密度大大增加,可以存储的信息量自然也比PMR硬盘明显更多。当然,极限操作毕竟不像常规操作那样稳妥,SMR技术下,磁盘可以存储的信息量大大增加了,但是缺点也很明显。首先是磁盘上的信息变得如此高密度,转速自然也不宜太快。所以SMR硬盘的转速一般都不快。其次就是,对于SMR硬盘而言,单纯的读写看起来很OK,但是如果想要修改某个磁道上的数据就比较麻烦了,因为磁道间隙比较小,而磁头比较宽,这样例如修改2磁道的数据,就必然会影响相邻的3磁道的数据。解决这个问题有两个途径,一个是每重叠一部分磁道时,隔开一些距离,另一个就是设置一些专用的缓冲区,当修改2磁道的数据时,先把3磁道的数据取出来放到缓冲区中,等2磁道的数据改完了,再将3磁道的数据放回去。看起来是一个很复杂的过程,所以SMR硬盘通常都具有一个特点:大缓存,一般能达到256MB的缓存,而普通PMR硬盘的缓存通常只有64MB。也正是由于这个过程比较复杂,所以在修改处理大量数据的时候会比较慢,时间久了对硬盘的读写性能会造成影响,甚至影响硬盘的寿命,造成数据损坏丢失等问题。所以,相较于PMR的硬盘,SMR硬盘是不适合用来当做系统盘或者需要频繁读写的硬盘来用的,它更适合当做小编在开头所说的仓储盘来使用,用来备份、留存一些数据。尽管现在硬盘的整体寿命已经有了很大的提升,但是当你要选购硬盘作为计算机主力硬盘时,还是应该尽可能避免买到SMR硬盘。3、如何区分自己的硬盘是PMR还是SMR?不过,比较尴尬的是,目前硬盘企业在产品包装上基本上是不会告诉你这块硬盘采用的是PMR还是SMR技术的,这就需要我们自己去辨别。网上很多小伙伴根据自己的自身经历以及经验常识,整理了一些方法来帮助大家辨别,这些方法只能作为参考,并不能百分百确定硬盘是PMR还是SMR技术。IT之家小编认为,最好的方法还是尽可能联系硬盘所属品牌的官方客服进行询问,这样得到的答案更为准确。当然如果你实在联系不上客服,那么小编也将网友整理的方法列在下方,供大家参考。1、看容量。SMR是为了追求硬盘容量而产生的方案,所以SMR硬盘的容量一般是比较大的。通常来说,3.5寸硬盘大于1TB,或者2.5寸硬盘大于500GB的,就有可能是SMR硬盘了。2、看缓存。刚才我们说到,SMR的技术特点导致它的缓存通常比较大,通常是128MB起步的。不过这个也不是定数,也有些SMR硬盘产品缓存比较小,只有64MB,但很少见,当然也有一些高端的PMR硬盘容量很大,缓存也能达到256MB。3、还有一个办法是根据硬盘的总容量计算每片磁盘的容量,硬盘的磁盘片数大家需要到对应品牌官网上去查找技术文档,如果这个品牌的产品没有提供技术文档,也可以寻找官方的客服解决。当然,如果你联系上了客服,或许可以直接询问该产品是PMR还是SMR盘了。当你了解了每碟磁盘的容量时,可以大概估摸硬盘是SMR还是PMR了。通常2.5寸一般每碟是500G左右,大的也能到ITB,而3.5寸一般是1TB左右,大的话有1.5TB。4、最后,Chiphell论坛有网友整理了市售3.5英寸SATA HDD的技术参数(点此前往),涵盖了目前市面上绝大部分的3.5英寸机械硬盘,其中就有硬盘是采用PMR技术还是SMR技术的信息,这份列表也可供大家参考。
文章推荐
智能摘要
延伸阅读
聊天咨询
责编:
来源:IT之家
#PMR#
#SMR#
收藏
点赞
分享至:
微信扫一扫分享
THE END
相关推荐
喜报 深圳芯佰特微电子有限公司 荣任深圳市无人机行业协会会员单位
帝奥微推出业界领先的电源及信号链产品系列DIO62820/DIO141X/DIO7610
和而泰:公司与小米已有业务合作,项目进展顺利
金百泽:基于英伟达Jetson AGXX avier评估板项目在开展中
深天马A:全制程产线预计今年具备小批量出货能力
大陆集团:2024年营收目标为410亿欧元至440亿欧元
+关注
爱集微
微信:
邮箱:laoyaoba@gmail.com
8.6w文章总数
12012.5w总浏览量
最近发布
武汉新芯“一种存储器件的制造方法及存储器件”专利公布
3小时前
京东方“显示基板和显示面板”专利获授权
3小时前
华为“存储阵列及其制备方法、存储器、电子设备”专利公布
3小时前
华为“一种强化学习的训练方法及相关装置”专利公布
3小时前
南通海门区“走进海门系列招商活动(深圳站)”即将开启 盛邀业界共谋发展
4小时前
获取更多内容
最新资讯
帝奥微推出业界领先的电源及信号链产品系列DIO62820/DIO141X/DIO7610
17分钟前
喜报 深圳芯佰特微电子有限公司 荣任深圳市无人机行业协会会员单位
17分钟前
和而泰:公司与小米已有业务合作,项目进展顺利
2小时前
金百泽:基于英伟达Jetson AGXX avier评估板项目在开展中
2小时前
深天马A:全制程产线预计今年具备小批量出货能力
3小时前
大陆集团:2024年营收目标为410亿欧元至440亿欧元
3小时前
PDF 加载中...
网站首页
版权声明
集微招聘
联系我们
网站地图
关于我们
商务合作
rss订阅
联系电话:
0592-6892326
新闻投稿:
laoyaoba@gmail.com
商务合作:
chenhao@ijiwei.com
问题反馈:
1574400753 (QQ)
官方微信
官方微博
APP下载
友情链接:
凤凰科技
雷锋网
财联社
电子产品世界
与非网
Copyright 2007-2023©.com™Inc.All rights reserved |
闽ICP备17032949号
闽公网安备 35020502000344号
CMR and SMR Hard Drives | Seagate ASEAN
CMR and SMR Hard Drives | Seagate ASEAN
Resource Centre
Blog
Open Source
Partners
Log-in
My Account
Search
View All Results
Support
Products
Knowledge Base
Support Downloads
Articles
suggested searches
Products
Solutions
Innovation
Support
About
Menu
Products
Cloud, Edge & Data Centre
Specialised Drives
Personal Storage
Data Storage Systems
Data Protected JBODs
Hybrid Storage Arrays
Expansion Enclosures and JBODs
Integrated Storage Servers
Enterprise Drives
Exos E Series
Exos X Series
Exos Mozaic 3+
Nytro SATA SSD Series
Nytro SAS SSD Series
Nytro NVME SSD Series
Lyve: Edge-to-Cloud Mass Storage Platform
Overcome the cost and complexity of storing, moving, and activating data at scale.
Lyve Cloud: Object Storage Designed for Multicloud
Lyve Mobile: Data Transfer as a Service
Lyve Mobile Shuttle
Lyve Mobile Array
Enhanced Reliability and Hands-Off Performance
Learn More
Gaming Hard Drives & SSDs
Find reliable gaming drives for PlayStation, Xbox and PC.
Xbox
PlayStation
PC Gaming
Special Edition Drive
NAS
IronWolf Hard Drives
IronWolf Pro Hard Drive
Video Analytics
SkyHawk Hard Drive
SkyHawk AI Hard Drive
External Drives
Ultra Touch HDD
One Touch HDD
One Touch SSD
One touch Hub
Expansion Portable
Expansion Desktop
Expansion SSD
Backup Plus Slim
Backup Plus Portable
Basic
Photo Drive
Internal Drives
BarraCuda 2.5" Hard Drive
BarraCuda 3.5" Hard Drive
BarraCuda SATA SSD
BarraCuda PCIe SSD
BarraCuda 515 M.2 NVMe SSD
BarraCuda 520 SSD
It's Mini. It's Mine
Learn More
Solutions
By Use Case
By Environment
By Partner
By Industry
Backup & Recovery
Safeguard business continuity with modern data protection
Big Data Analytics (AI/ML)
Power your analytics with storage optimised for mass capacity
Cloud Backup
Activate backups and improve data security, efficiency and compliance
Data Migration
Extract more value from legacy content with managed migration services
Data Transfer
The right-sized approach to any data transfer
Edge
Deploy cost effective storage and data workflow solutions at the edge
High Performance Computing (HPC)
Maximise performance to meet data-intensive workloads
Multicloud
Transfer, store, & access data across multicloud environments
Private Cloud
Significantly reduce TCO for secure, scalable cloud storage
Public Cloud
Data storage solutions with cloud flexibility and cost predictability
Storage as a Service
A frictionless cloud, available at the metro edge
Video Analytics
Unlock video and analytics with valuable insights from edge to cloud
Enhanced Reliability and Hands-Off Performance
Learn More
Edge
Deploy cost effective storage and data workflow solutions at the edge
Multicloud
Transfer, store, & access data across multicloud environments
Private Cloud
Significantly reduce TCO for secure, scalable cloud storage
Public Cloud
Data storage solutions with cloud flexibility and cost predictability
Enhanced Reliability and Hands-Off Performance
Learn More
Featured Partners
Commvault
Veeam
VMWare
Milestone
Equinix
EVS
NI
IBM
View All Seagate Technology Partners
Exclusive Hybrid Edge-to-Cloud Solution Bundles
Learn More
Autonomous Vehicles
Massive in-vehicle storage capacity and modular edge solutions
Healthcare
Effectively manage and orchestrate life sciences data
Media & Entertainment
Streamline content workflows and efficiently store media data
Surveillance & Security
Unlock valuable surveillance insights from edge to cloud
Telecommunications
Meet high-speed network demands with exabyte scalability
Geosciences & Energy
Accelerate geophysical data delivery with storage that’s built for the edge.
Enhanced Reliability and Hands-Off Performance
Learn More
Multicloud Maturity Report
Find Out How Your Business Can Thrive in the Multicloud
Innovation
Our Innovations
Mozaic 3+ Platform
Hard drive platform for unprecedented areal density
MACH.2 Multi-Actuator Hard Drives
Multi-Actuator Technology
Lyve Labs
A program for startups and innovators.
Seagate Secure
Delivering reliable, world-class data protection solutions
Open-Source Developer Community
Creating accessible storage solutions through collaboration
Composable Infrastructure
Dynamically allocate and configure resources to any application process
Seagate Circularity Program
We build our hard drives to last. Give yours a second life.
Enhanced Reliability and Hands-Off Performance
Learn More
Support
All Support Options
SUPPORT HOME
Seagate Support Home - Find important support related documentation, Seagate compliance documents, popular downloads, browse our top support articles
Products
Find product-specific documentation, knowledge base articles, videos and other self-service tools
Software Downloads
Browse and download the latest software, apps, utilities, plug-ins and content.
Warranty & Replacements
Check your drive's warranty status and browse our warranty related documentation.
Seagate Systems
Find documentation, articles and other self-service tool for Seagate Systems
Lyve Mobile
Find documentation, articles, videos, and other self-service tools for high-capacity edge storage and mass data transfer
Lyve Cloud
Looking for Help documentation on Lyve Cloud or need support? Browse our Lyve Cloud support articles.
Product Registration
Register your Seagate product(s).
การสนับสนุนสินค้า
Hỗ trợ Sản phẩm tại Việt Nam
About
About Seagate
Our Story
Learn about the legacy we’re building one terabyte at a time
Leadership Team
Board of Directors
ESG
Trust Centre
Quality Assurance
Join our Seagate Team
Are you creative, collaborative, and passionate? Join us.
Life at Seagate
Diversity, Equity and Inclusion
Employee Resource Groups
University Programmes
Investors
Learn the latest — from upcoming events to stocks, financials and more.
Press Centre
Access the news and resources you need.
News
Media Kits
Brand Portal
Read Now
Products
Solutions
Innovation
Support
About
Resource Centre
Blog
Open Source
Partners
Log-in
My Account
Main
Products
Cloud, Edge & Data Centre
Specialised Drives
Personal Storage
Main
Solutions
By Use Case
By Environment
By Partner
By Industry
Main
Innovation
Our Innovations
Main
Support
All Support Options
Main
About
About Seagate
Products
Cloud, Edge & Data Centre
Data Storage Systems
Data Protected JBODs
Hybrid Storage Arrays
Expansion Enclosures and JBODs
Integrated Storage Servers
Enterprise Drives
Exos E Series
Exos X Series
Exos Mozaic 3+
Nytro SATA SSD Series
Nytro SAS SSD Series
Nytro NVME SSD Series
Lyve: Edge-to-Cloud Mass Storage Platform
Lyve Cloud: Object Storage Designed for Multicloud
Lyve Mobile: Data Transfer as a Service
Lyve Mobile Shuttle
Lyve Mobile Array
Products
Specialised Drives
Gaming Hard Drives & SSDs
Xbox
PlayStation
PC Gaming
Special Edition Drive
NAS
IronWolf Hard Drives
IronWolf Pro Hard Drive
Video Analytics
SkyHawk Hard Drive
SkyHawk AI Hard Drive
Products
Personal Storage
External Drives
Ultra Touch HDD
One Touch HDD
One Touch SSD
One touch Hub
Expansion Portable
Expansion Desktop
Expansion SSD
Backup Plus Slim
Backup Plus Portable
Basic
Photo Drive
Internal Drives
BarraCuda 2.5" Hard Drive
BarraCuda 3.5" Hard Drive
BarraCuda SATA SSD
BarraCuda PCIe SSD
BarraCuda 515 M.2 NVMe SSD
BarraCuda 520 SSD
Solutions
By Use Case
Backup & Recovery
Big Data Analytics (AI/ML)
Cloud Backup
Data Migration
Data Transfer
Edge
High Performance Computing (HPC)
Multicloud
Private Cloud
Public Cloud
Storage as a Service
Video Analytics
Solutions
By Environment
Edge
Multicloud
Private Cloud
Public Cloud
Solutions
By Partner
Featured Partners
Commvault
Veeam
VMWare
Milestone
Equinix
EVS
NI
IBM
View All Seagate Technology Partners
Solutions
By Industry
Autonomous Vehicles
Healthcare
Media & Entertainment
Surveillance & Security
Telecommunications
Geosciences & Energy
Innovation
Our Innovations
Mozaic 3+ Platform
MACH.2 Multi-Actuator Hard Drives
Lyve Labs
Seagate Secure
Open-Source Developer Community
Composable Infrastructure
Seagate Circularity Program
Support
All Support Options
SUPPORT HOME
Products
Software Downloads
Warranty & Replacements
Seagate Systems
Lyve Mobile
Lyve Cloud
Product Registration
การสนับสนุนสินค้า
Hỗ trợ Sản phẩm tại Việt Nam
About
About Seagate
Our Story
Leadership Team
Board of Directors
ESG
Trust Centre
Quality Assurance
Join our Seagate Team
Life at Seagate
Diversity, Equity and Inclusion
Employee Resource Groups
University Programmes
Investors
Press Centre
News
Media Kits
Brand Portal
Select Your Country/Location & Language
Remember my country/location & language selection
Asia-Pacific (APAC)
ASEAN (English)
Australia/New Zealand (English)
India (English)
Indonesia (Indonesian)
Singapore (English)
대한민국 (한국어)
日本 (日本語)
台灣 (繁體中文)
中国 (简体中文)
Europe
België (Nederlands)
Belgique (Français)
Deutschland (Deutsch)
España (Español)
France (Français)
Italia (Italiano)
Nederland (Nederlands)
Polska (Polski)
Portugal (Português)
United Kingdom (English)
Middle East/North Africa
Middle East/North Africa (English)
Türkiye (Türkçe)
Latin America
Brasil (Português)
Latinoamérica (Español)
North America
Canada (English)
Canada (Français)
United States (English)
Log in
Seagate Partner Program Portal
Provides access to product training, sales and marketing resources, deal registration, and more to our VARs, Integrators, Resellers and other channel partners.
Lyve Cloud Portal
Use the Lyve Cloud portal to configure and manage your object storage and services.
Seagate Direct Customer Portal
Provides Direct customers with B2B Self Service tools such as Pricing, Programs, Ordering, Returns and Billing.
Lyve Management Portal
Register, access, and manage Lyve Mobile services, subscriptions and projects.
Seagate Supplier Portal
Provides Suppliers with self-service tools targeted to the needs of their business.
Consumer Portal
View registered products, register new products, and find product specific support.
My Profile
View/edit your profile information
My Dashboard
View your dashboard
Log out
Logout of your account
CMR and SMR Hard Drives
Which Drive Has What?
Our drives are purpose-built, meaning each of our internal drives are built specifically for their intended use. Each drive has been optimised, tested, and approved by various technology partners to meet and exceed the requirements for its application in terms of reliability, performance and quality. Seagate drives utilise conventional magnetic recording and shingled magnetic recording technologies based on intended use. Below is a table of our internal drives with each of their respective recording technology.
3.5-inch Drives1,2
EXOS X
CMR12 TB, 14 TB, 16 TB, 18 TB, 20 TB
SMR-
Mozaic HAMR24, 30 TB
EXOS E
CMR1 TB, 2 TB, 3 TB, 4 TB, 6 TB, 8 TB, 10 TB
SMR-
Mozaic HAMR-
IronWolf
CMR1 TB, 2 TB, 3 TB, 4 TB, 6 TB, 8 TB, 10 TB+
SMR-
Mozaic HAMR-
IronWolf Pro
CMR2 TB, 4 TB, 6 TB, 8 TB, 10 TB+
SMR-
Mozaic HAMR24 TB
BarraCuda
CMR1 TB
SMR2 TB, 3 TB, 4 TB, 5 TB, 6 TB, 8 TB
Mozaic HAMR-
BarraCuda Pro
CMR2 TB, 4 TB, 6 TB, 8 TB, 10 TB+
SMR-
Mozaic HAMR-
FireCuda
CMR1 TB, 2 TB
SMR-
Mozaic HAMR-
Archive
CMR-
SMR8 TB
Mozaic HAMR-
SkyHawkLite/Mini
CMR-
SMR1 TB, 2 TB
Mozaic HAMR-
SkyHawk
CMR1 TB, 2 TB, 3 TB, 4 TB, 6 TB, 8 TB
SMR-
Mozaic HAMR-
SkyHawk AI
CMR8 TB, 10 TB+
SMR-
Mozaic HAMR-
2.5-inch Drives1,2
BarraCuda
SMR500 GB, 1 TB, 2 TB, 3 TB+
FireCuda
SMR1 TB, 2 TB
SkyHawkLite/Mini
SMR1 TB, 2 TB
EXOS E
CMR300 GB, 600 GB, 1 TB, 1.2 TB, 1.8 TB, 2 TB, 2.4 TB
Mozaic 3+: Where the Future is Read and Written
Mozaic 3+ is a hard drive platform that incorporates Seagate’s unique implementation of heat-assisted magnetic recording (HAMR) technology to deliver mass-capacity storage at unprecedented areal densities of 3 TB/disk and beyond.
Mozaic 3+ is a composite of the most complex nanoscale recording technologies and material science breakthroughs on the planet and is how data can now be stored to media at density levels that were previously inconceivable, all while using the same material resources as current Seagate hard drives and delivered in a known 3.5-inch form factor. Mozaic 3+ is both the present and future, with an eye toward 50 TB and still “10 platters and 20 heads”.
With Mozaic 3+, data centre operators can store more exabytes in less space, with fewer racks, amounting to massive TCO savings, with energy consumed and floor space.
Learn More: https://www.seagate.com/innovation/mozaic.
1. Reflects products available through Seagate authorised distributors/resellers2. Data may change, please refer to detailed product information available at Seagate.com
Multicloud Maturity Report
Find Out How Your Business Can Thrive in the Multicloud
About Seagate
Our Story
ESG
Trust Centre
Quality Assurance
Support
Product Support
Software Downloads
Warranty & Replacements
Seagate Systems
Contact Us
Press Centre
News
Media Kits
Brand Portal
Investors
Partners
Indirect Partners & Resellers
Seagate Training Portal
Seagate Direct & Suppliers
Seagate Technology Alliance Program
Jobs
Life at Seagate
Diversity, Equity and Inclusion
University Programmes
Where to Buy
Product Finder
©2024 Seagate Technology LLC
Legal
Privacy
Vulnerability Disclosure
Cookie Settings
LaCie
Maxtor
Lyve Labs
Seagate Government Solutions
Site Map